
Infective Endocarditis in High-Income Countries

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Special Populations of Infectious Endocarditis in the 21st Century
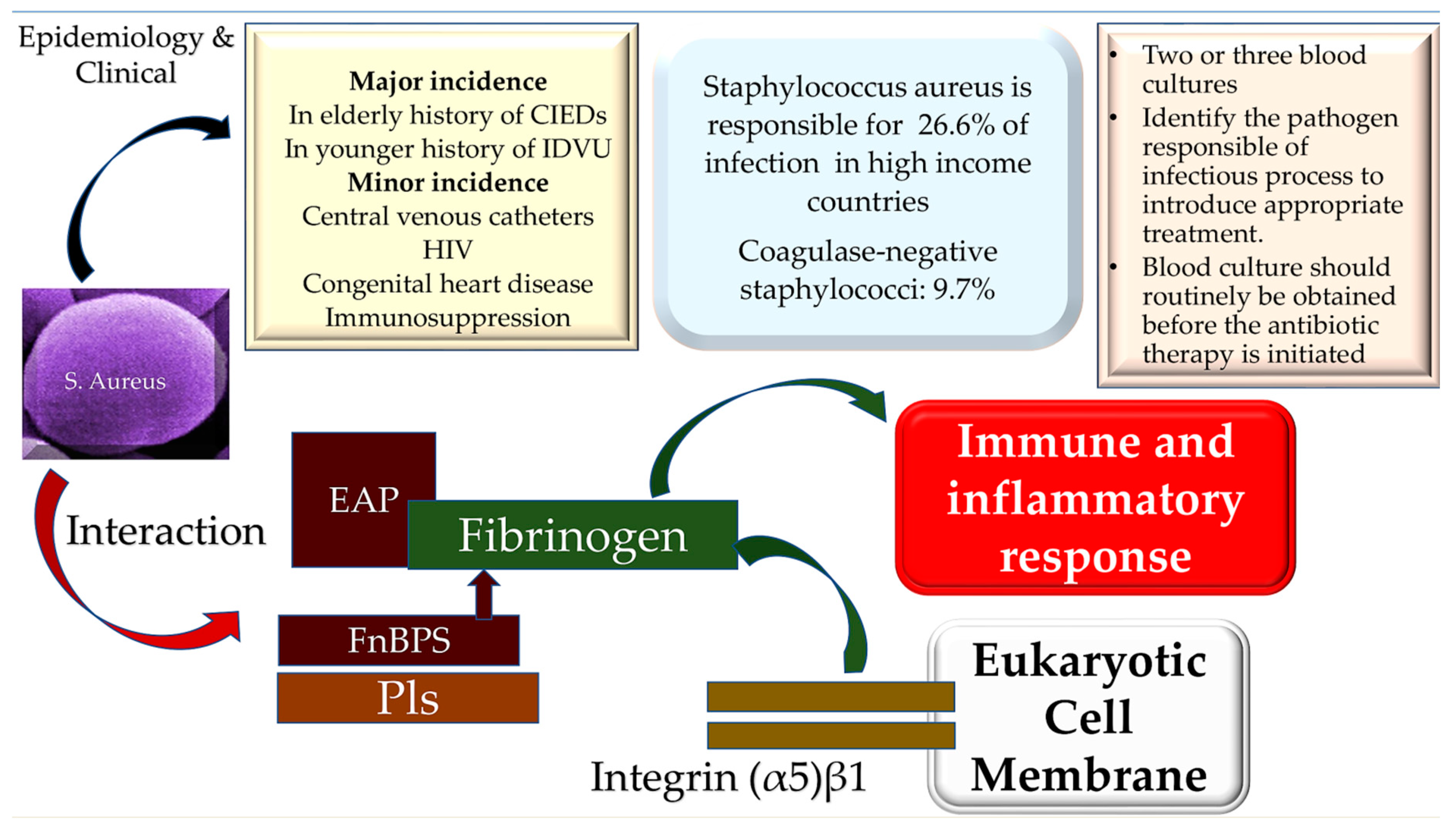
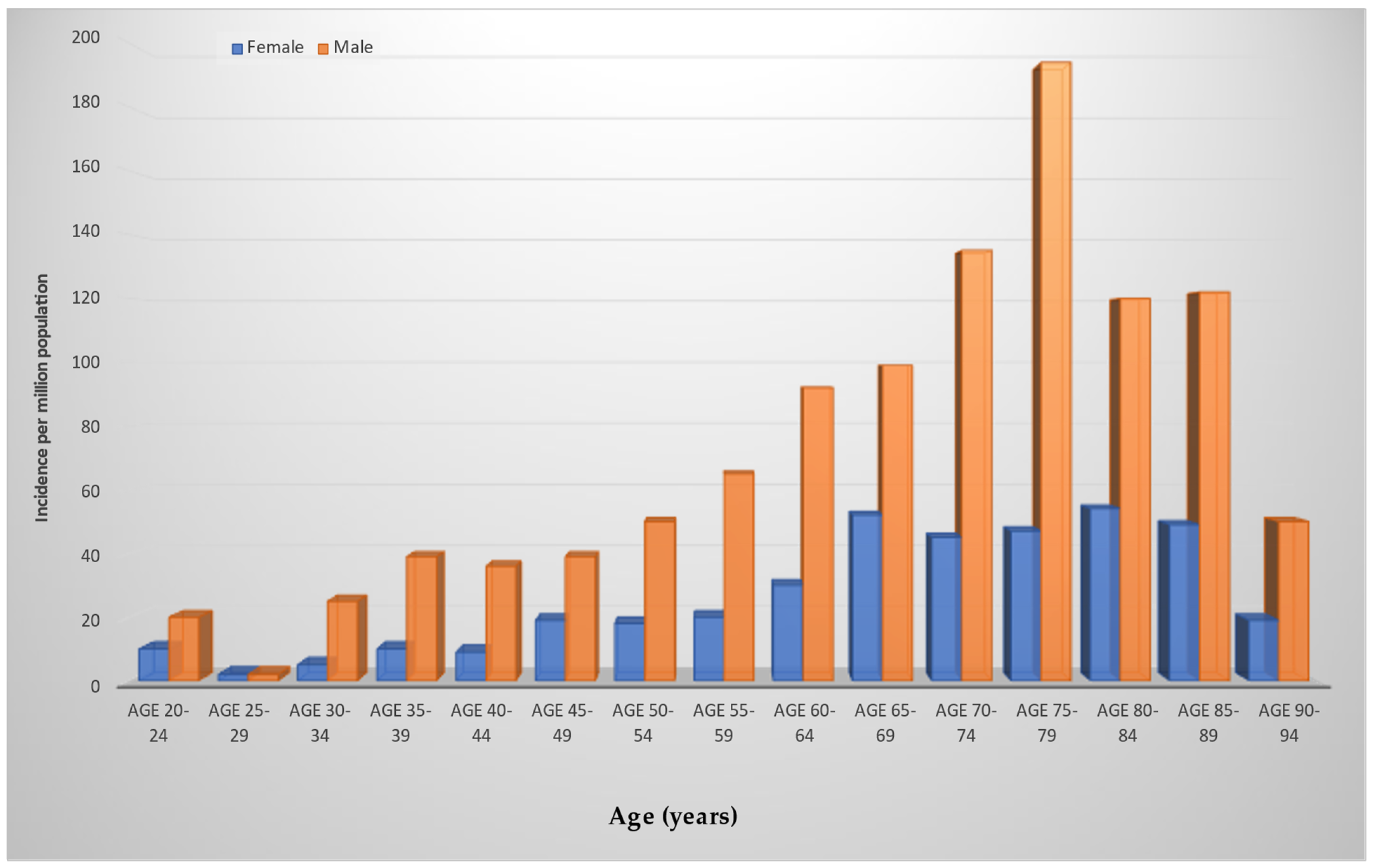
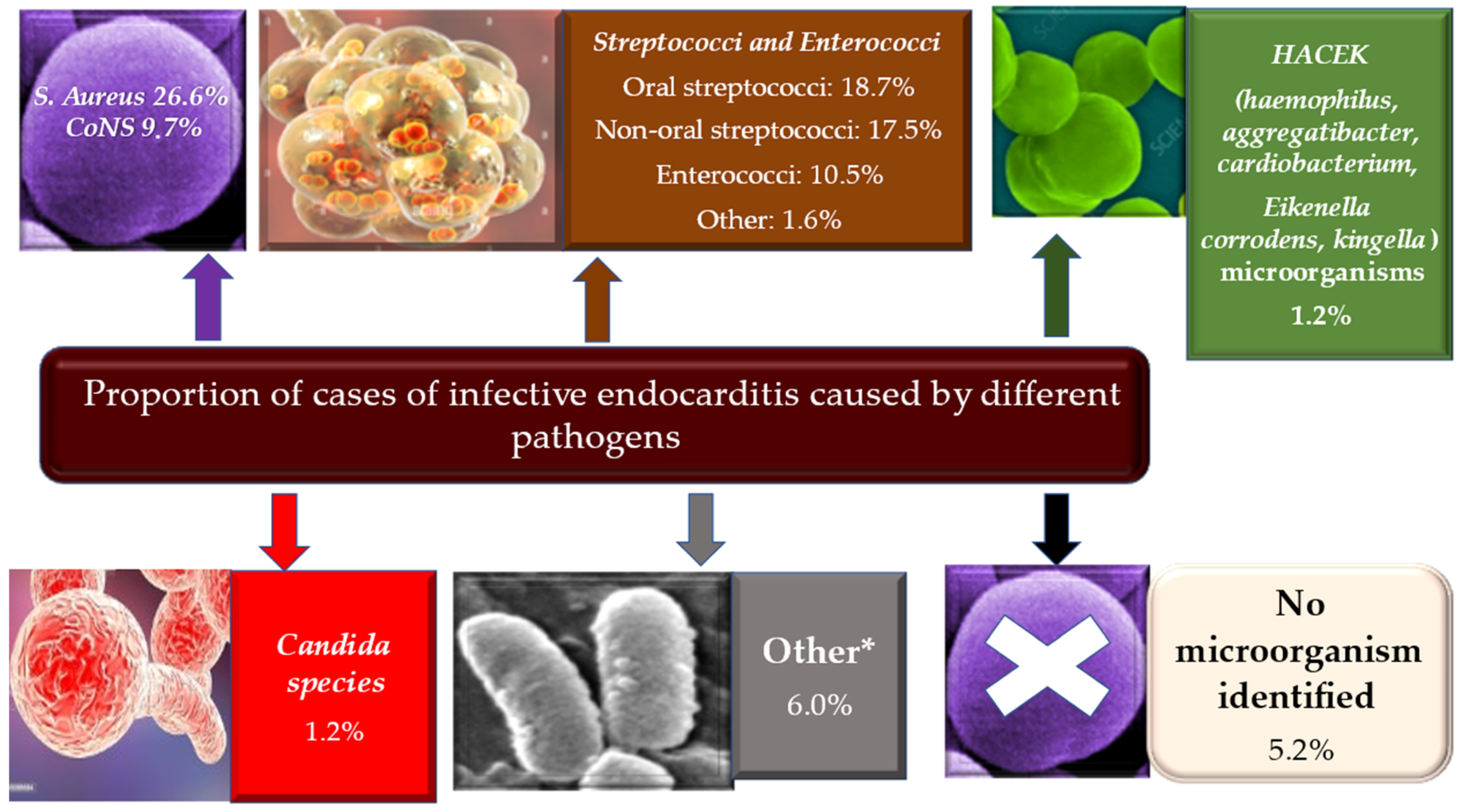
3. Epidemiology
4. Microbiology
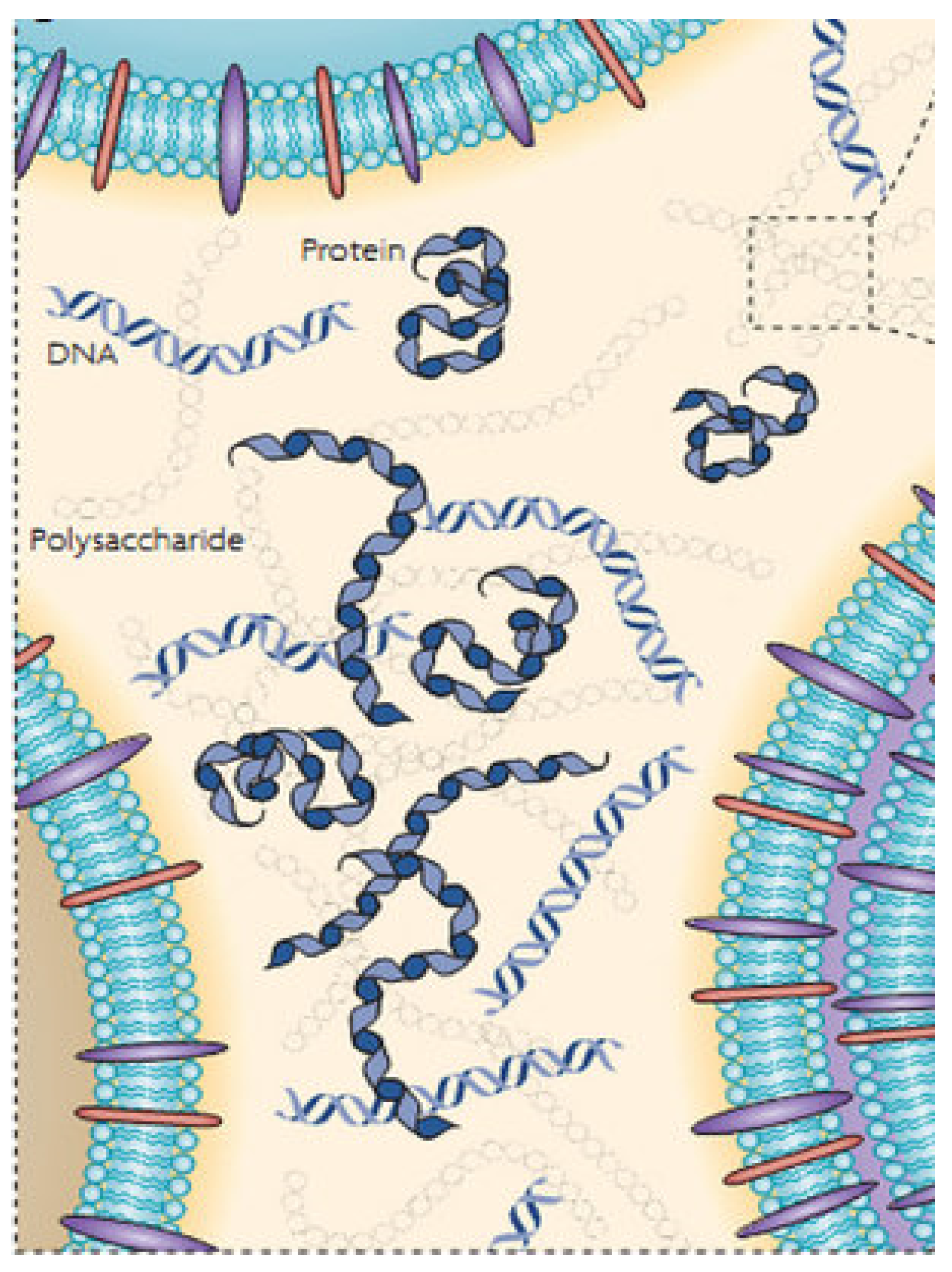
4.1. Biofilm Formation
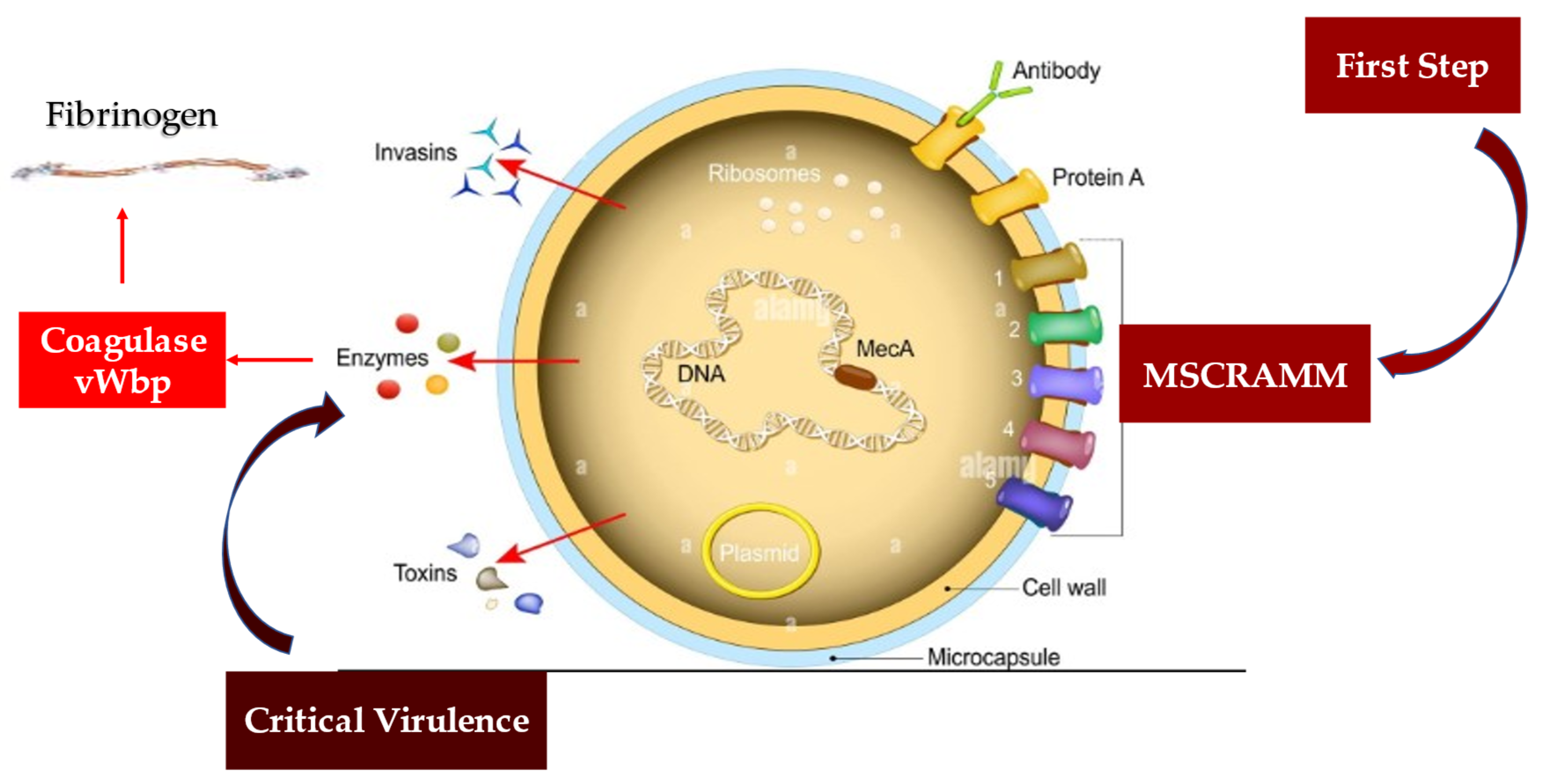
4.2. Staphylococcus aureus Protective Shield and Host Protection Mechanisms: New Evidence from Infectious Deployment
5. Pathophysiology
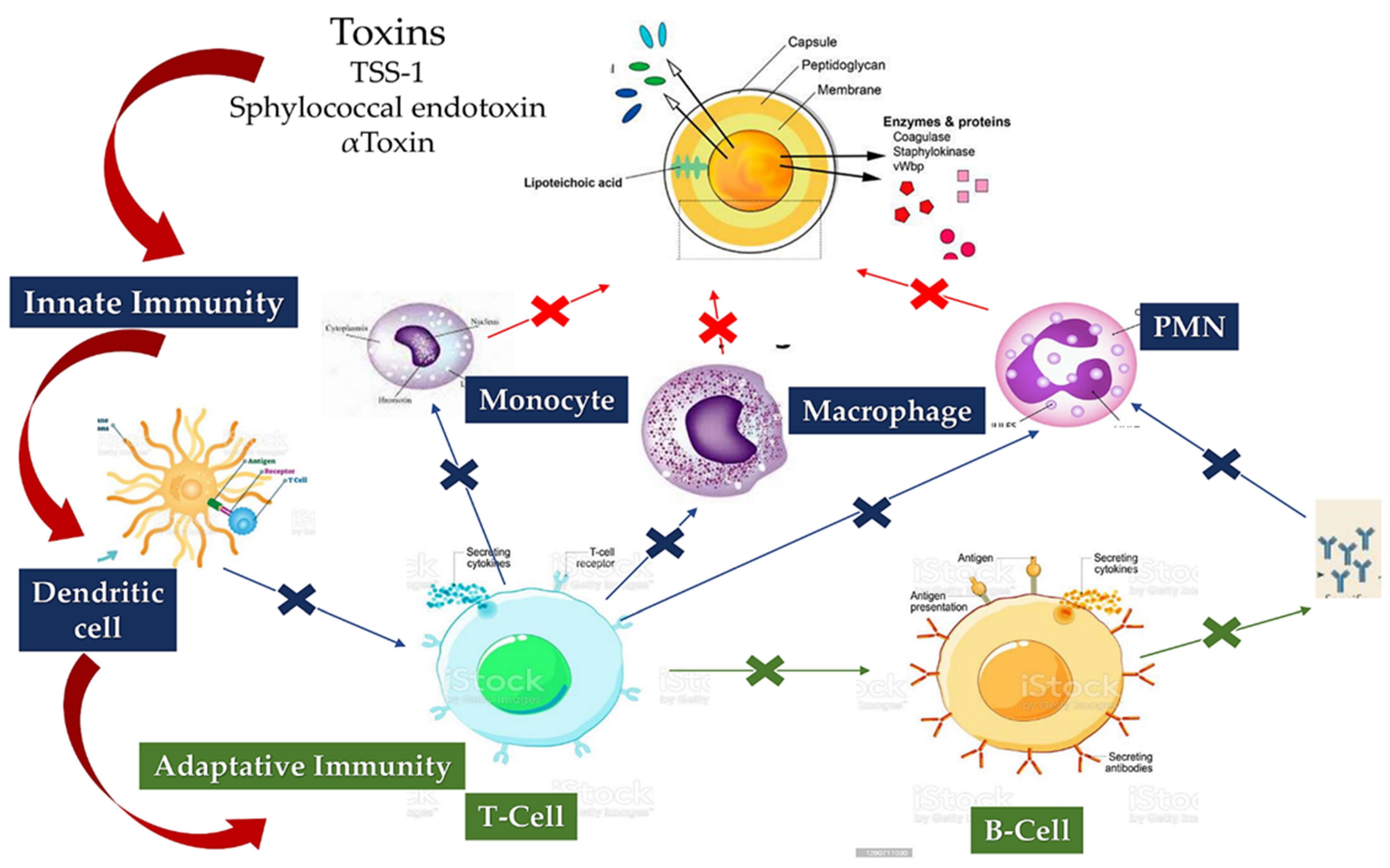
5.1. Staphylococcus aureus Immunity
5.1.1. Staphylococcus aureus Interacts with Host Innate Immunity
5.1.2. Staphylo Cytotoxins Are a Trojan Horse for Excellent Immune Modulation
5.1.3. Loss of B-Cell vs. T-Cell Cooperation due to Cumulative Effects of B-Cell Deletion and Lack of T-Cell Help
5.1.4. Immunoresponse and Vaccine
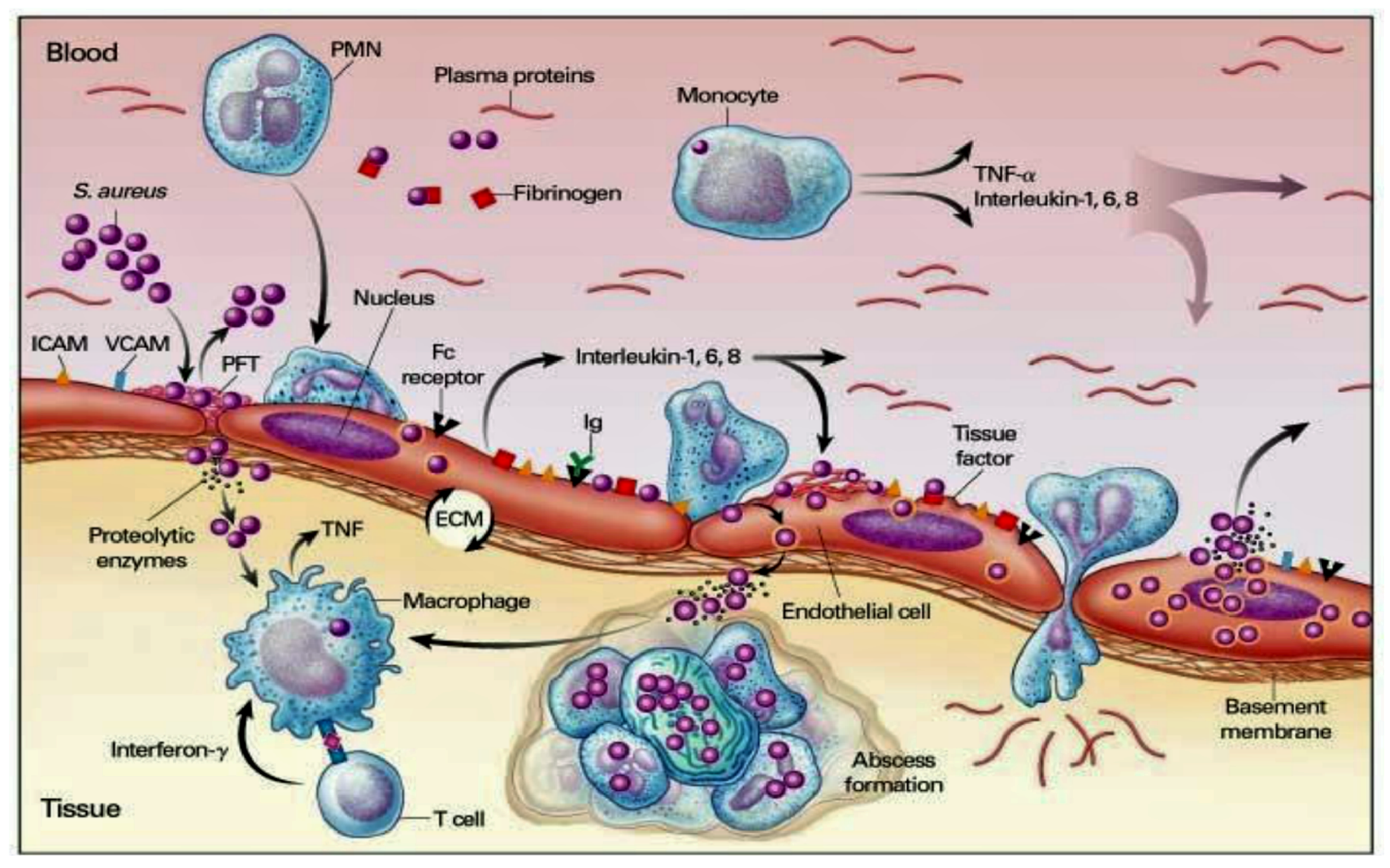
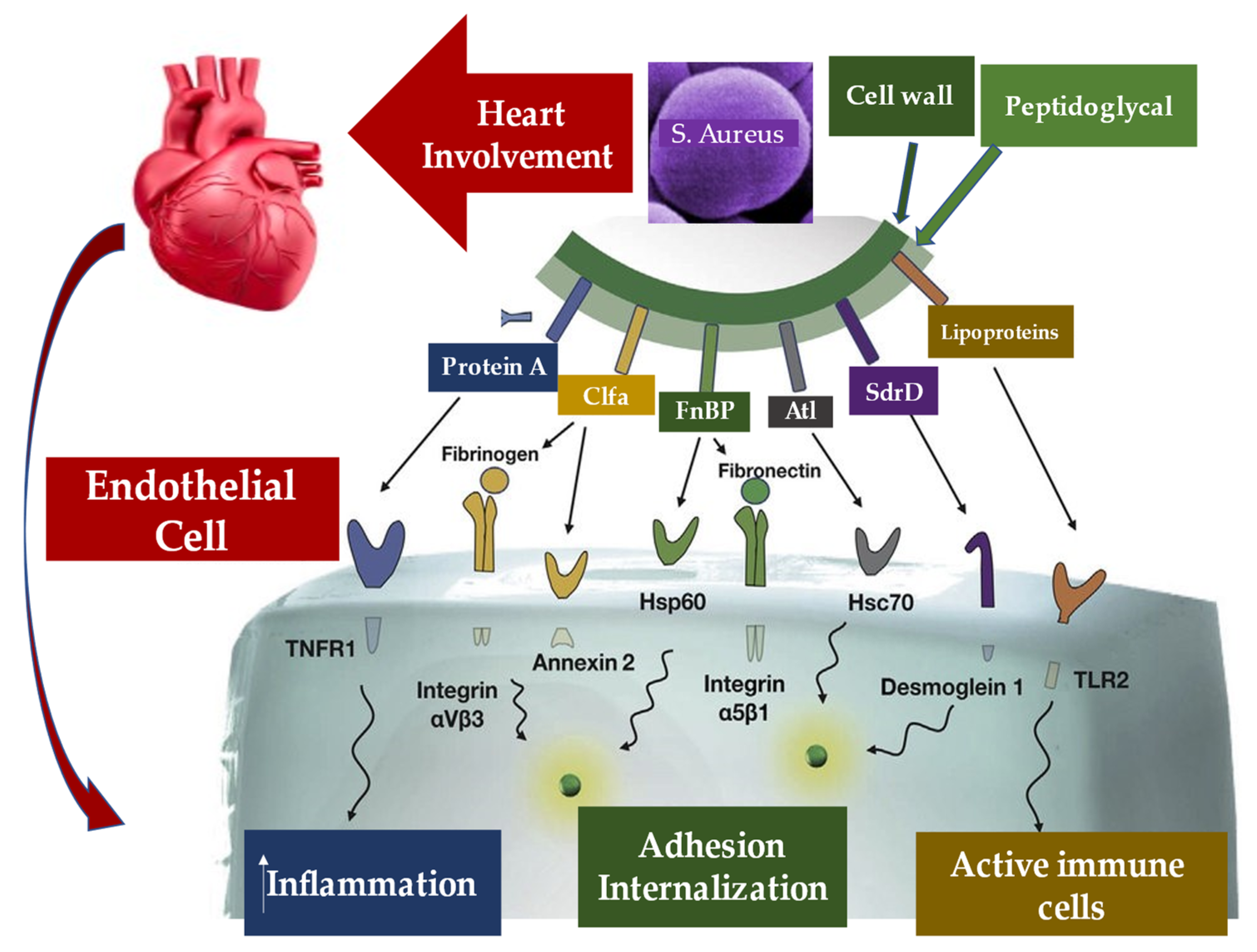
5.2. Pathogen–Host Interaction in Determining Inflammation
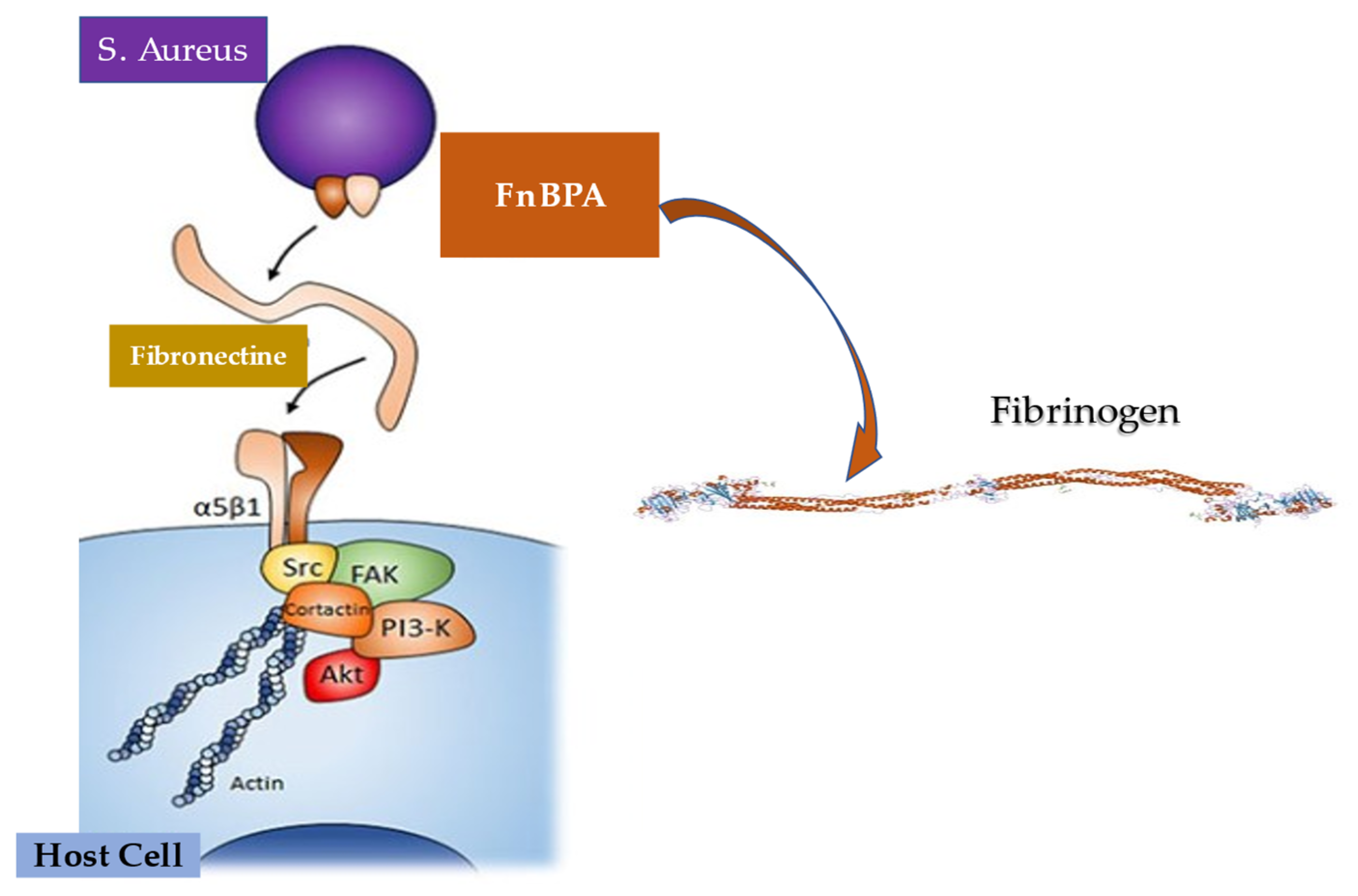
5.3. Interaction between Infective Endocarditis Pathogens, Vascular Endothelium, and Blood Constituents
5.4. Infective Endocarditis and Platelets
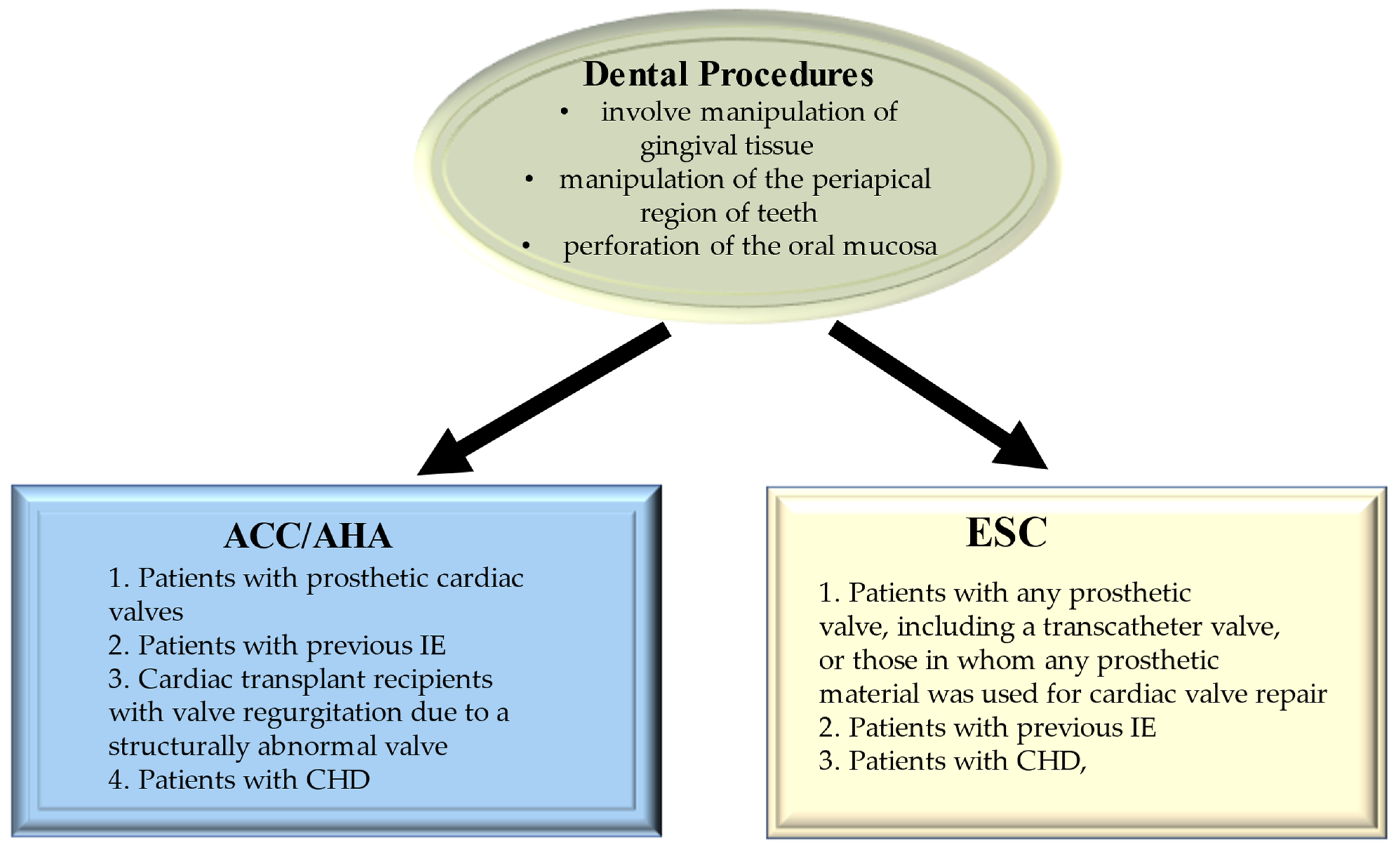
6. Evidence from Deploying Maneuvers as a Risk Factor for Bacteremia Related to Infective Endocarditis
6.1. Special Population Requiring Attention
6.1.1. Tooth Extraction and Tooth Brushing
6.1.2. Causative Pathogens of Interest and Related Mechanism Leading to Disease
6.2. Cardiac Device Infection
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAM | A disintegrin and metalloprotease |
| ADAMTS13 | A disintegrin and metalloprotease with thrombospondin type 1 repeats |
| Agr | Accessory Gene Regulatory System |
| Atl | autolysin |
| CHD | congenital heart disease |
| CIED | cardiac implantable electronic devices |
| Clf | cell-bound clumping factor |
| Coa | coagulase |
| CoNS | coagulase negative |
| EC | endothelial cell |
| Efb1 | extracellular fibrinogen binding protein |
| EPS | extracellular polymeric substances |
| ESAT-6 | Early Secreted Antigenic Target 6 kDa |
| Fg | fibrinogen |
| Fnb | fibronectin binding protein |
| HIV | immunodeficiency virus |
| Hla | human leukocyte antigen |
| Hsc70 | Heat shock cognate |
| Hsp60 | Heat shock protein |
| KC | keratinocyte |
| ICAM | Inter Cellular Adhesion Molecule |
| IE | Infective endocarditis |
| IL | interleukine |
| LAVD | left ventricular assist device |
| LC3-II | light chain 3 type II |
| Luk | leukotoxins |
| MntC | manganese transport protein C |
| MSCRAMM | microbial surface components recognizing adhesive matrix molecules |
| NK | natural killers |
| NLR | Nod-Like receptor |
| NLRP3 | NOD-like receptor family pyrin domain-containing 3 |
| PMN | polymorphonuclear |
| PVL | proviral load |
| PWID | persons who inject drugs |
| S aureus | Staphylococcus aureus |
| SpA | Staphylococcal Protein A |
| SdrD | Serine Aspartate repeat containing protein D |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TNFR1 | tumor necrosis factor receptor 1. |
| TSS-1 | Toxic Shock Syndrome-1 |
| ULVWF | Ultra Large vWF |
| United States | US |
| vWbp | von Willebrand factor-binding protein |
| vWF | von Willebrand factor |
| VCAM | vascular cell adhesion molecule |
References
- Murdoch, D.R.; Corey, G.R.; Hoen, B.; Miró, J.M.; Fowler, V.G.; Bayer, A.S.; Karchmer, A.W.; Olaison, L.; Pappas, P.A.; Moreillon, P.; et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: The International Collaboration on Endocarditis-Prospective Cohort Study. Arch. Intern. Med. 2009, 169, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Liaqat, W.; Palaiodimos, L.; Li, W.; Karamanis, D.; Tahir, A.; Tzoumas, A.; Nagraj, S.; Tiwari, N.; Grushko, M.; Kokkinidis, D.; et al. Epidemiologic and clinical characteristics of infective endocarditis: A single-center retrospective study in the Bronx, New York. Infection 2022, 1–13. [Google Scholar] [CrossRef]
- Paul, G.; Ochs, L.; Hohmann, C.; Baldus, S.; Michels, G.; Meyer-Schwickerath, C.; Fätkenheuer, G.; Mader, N.; Wahlers, T.; Weber, C.; et al. Surgical Procedure Time and Mortality in Patients with Infective Endocarditis Caused by Staphylococcus aureus or Streptococcus Species. J. Clin. Med. 2022, 11, 2538. [Google Scholar] [CrossRef]
- Selton-Suty, C.; Célard, M.; Le Moing, V.; Doco-Lecompte, T.; Chirouze, C.; Iung, B.; Strady, C.; Revest, M.; Vandenesch, F.; Bouvet, A. Preeminence of Staphylococcus aureus in infective endocarditis: A 1-year population-based survey. Clin. Infect. Dis. 2012, 54, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhan, Y.; Zhang, K.; Gao, Y.; Chen, L.; Zhan, J.; Chen, Z.; Zeng, Z. The Global, Regional, and National Burden and Trends of Infective Endocarditis From 1990 to 2019: Results from the Global Burden of Disease Study 2019. Front. Med. 2022, 9, 774224. [Google Scholar] [CrossRef] [PubMed]
- Resende, P., Jr.; Fortes, C.Q.; do Nascimento, E.M.; Sousa, C.; Fortes, N.R.Q.; Thomaz, D.C.; de Bragança Pereira, B.; Pinto, F.J.; de Oliveira, G.M.M. In-hospital Outcomes of Infective Endocarditis from 1978 to 2015: Analysis through Machine-Learning Techniques. CJC Open 2021, 4, 164–172. [Google Scholar] [CrossRef]
- Allegranzi, B.; Nejad, S.B.; Combescure, C.; Graafmans, W.; Attar, H.; Donaldson, L.; Pittet, D. Burden of endemic health-care-associated infection in developing countries: Systematic review and meta-analysis. Lancet 2011, 377, 228–241. [Google Scholar] [CrossRef]
- Bagheri Nejad, S.; Allegranzi, B.; Syed, S.B.; Ellis, B.; Pittet, D. Health-care-associated infection in Africa: A systematic review. Bull. World Health Organ. 2011, 89, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Joubert, D.; Cullati, S.; Briot, P.; Righi, L.; Grauser, D.; Ourahmoune, A.; Chopard, P. How to improve hospital admission screening for patients at risk of multidrug-resistant organism carriage: A before-and-after interventional study and cost-effectiveness analysis. BMJ Open Qual. 2022, 11, e001699. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Martínez, J.L. Antibiotics and antibiotic resistance genes in natural environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, J.; Wei, Y.; Zhang, J.; Tao, C. Recent advances in metal-organic framework-based materials for anti-staphylococcus aureus infection. Nano Res. 2022, 15, 6220–6242. [Google Scholar] [CrossRef]
- Prendergast, B.D. The changing face of infective endocarditis. Heart 2006, 92, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Habib, G.; Hoen, B.; Tornos, P.; Thuny, F.; Prendergast, B.; Vilacosta, I.; Moreillon, P.; Antunes, M.d.; Thilen, U.; Lekakis, J.; et al. Guidelines on the prevention, diagnosis, and treatment of infective endocarditis (new version 2009): The Task Force on the Prevention, Diagnosis, and Treatment of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by the European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and the International Society of Chemotherapy (ISC) for Infection and Cancer. Eur. Heart J. 2009, 30, 2369–2413. [Google Scholar] [PubMed] [Green Version]
- van den Brink, F.S.; Swaans, M.J.; Hoogendijk, M.G.; Alipour, A.; Kelder Johannes, C.; Jaarsma, W.; Eefting, F.D.; Groenmeijer, B.; Kupper, A.J.F.; ten Berg, J.M. Increased incidence of infective endocarditis after the 2009 European Society of Cardiology guideline update: A nationwide study in the Netherlands. Eur. Heart J. Qual. Care Clin. Outcomes 2017, 3, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Otto, C.M.; Nishimura, R.A.; Bonow, R.O.; Carabello, B.A.; Erwin, J.P., 3rd; Gentile, F.; Jneid, H.; Krieger, E.V.; Mack, M.; McLeod, C.; et al. 2020 ACC/AHA Guideline for the Management of Patients With Valvular Heart Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2021, 77, 450–500. [Google Scholar] [CrossRef]
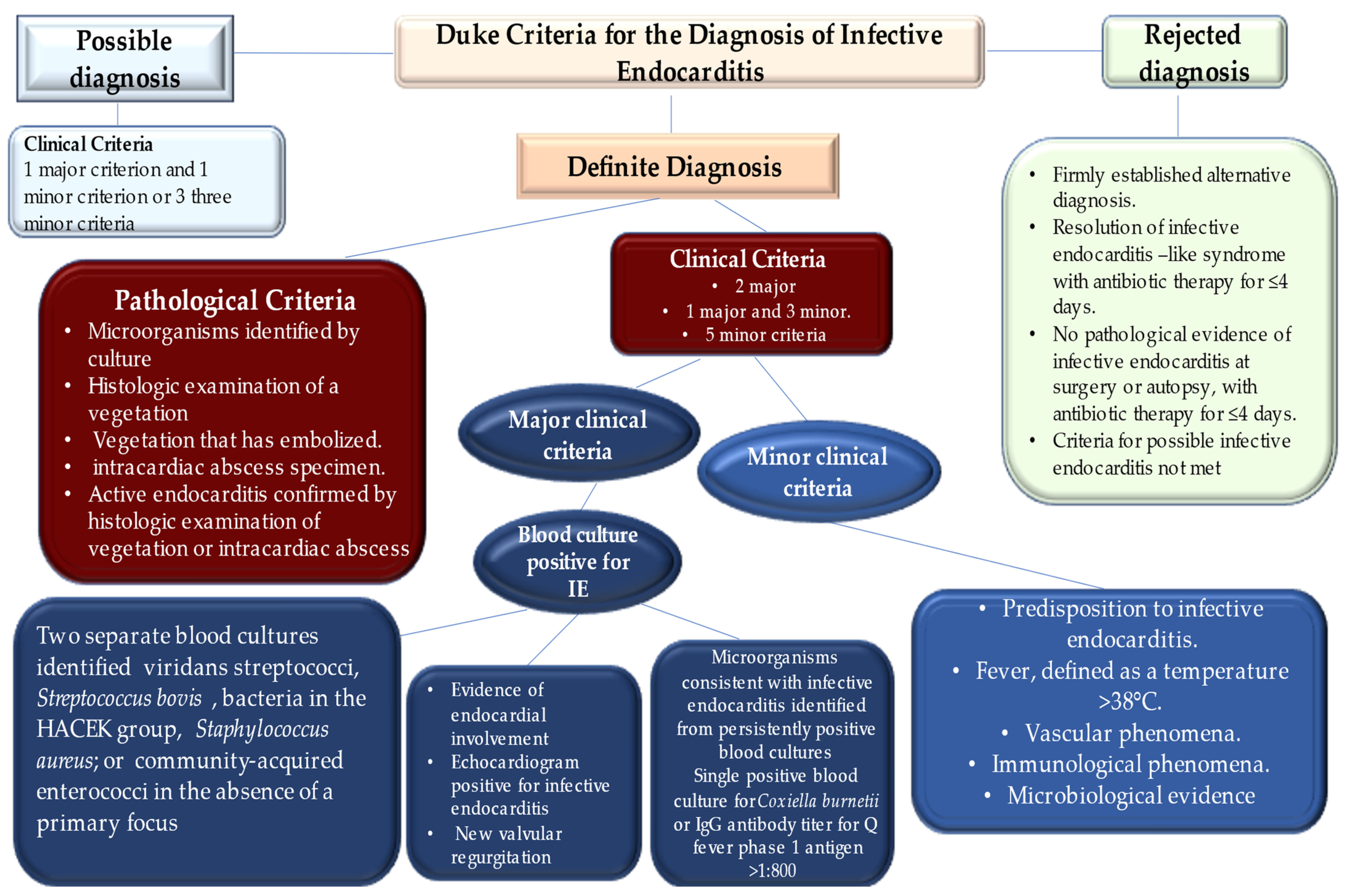
- Li, J.S.; Sexton, D.J.; Mick, N.; Nettles, R.; Fowler, V.G., Jr.; Ryan, T.; Bashore, T.; Corey, G.R. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin. Infect. Dis. 2000, 30, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Richet, H.; Casalta, J.P.; Thuny, F.; Mérrien, J.; Harlé, J.R.; Weiller, P.J.; Habib, G.; Raoult, D. Development and assessment of a new early scoring system using non-specific clinical signs and biological results to identify children and adult patients with a high probability of infective endocarditis on admission. J. Antimicrob. Chemother. 2008, 62, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Mahabadi, A.A.; Mahmoud, I.; Dykun, I.; Totzeck, M.; Rath, P.M.; Ruhparwar, A.; Buer, J.; Rassaf, T. Diagnostic value of the modified Duke criteria in suspected infective endocarditis -The PRO-ENDOCARDITIS study. Int. J. Infect. Dis. 2021, 104, 556–561. [Google Scholar] [CrossRef]
- Gouriet, F.; Tissot-Dupont, H.; Casalta, J.; Pm Hubert, S.; Fournier, P.E.; Edouard, S.; Theron, A.; Lepidi, H.; Grisoli, D.; Habib, G.; et al. Marseille scoring system for empiric treatment of infective endocarditis. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 841–849. [Google Scholar] [CrossRef]
- Crawford, M.H.; Durack, D.T. Clinical presentation of infective endocarditis. Cardiol. Clin. 2003, 21, 159–166. [Google Scholar] [CrossRef]
- Dabbous, M.; Saba, M.; El Orra, S.; Najjar, C. Confusion as an Unusual Presentation of Infective Endocarditis. Cureus 2021, 13, e20809. [Google Scholar] [CrossRef] [PubMed]
- Thuny, F.; Avierinos, J.F.; Tribouilloy, C.M.; Giorgi, R.; Casalta, J.P.; Milandre, L.; Brahim, A.; Nadji, G.; Riberi, A.; Collart, F.; et al. Impact of cerebrovascular complications on mortality and neurologic outcome during infective endocarditis: A prospective multicentre study. Eur. Heart J. 2007, 28, 1155–1161. [Google Scholar] [CrossRef] [Green Version]
- Sonneville, R.; Mirabel, M.; Hajage, D.; Tubach, F.; Vignon, P.; Perez, P.; Lavoué, S.; Kouatchet, A.; Pajot, O.; Mekontso Dessap, A.; et al. Neurologic complications and outcomes of infective endocarditis in critically ill patients: The ENDOcardite en REAnimation prospective multicenter study. Crit. Care Med. 2011, 39, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Dickerman, S.A.; Abrutyn, E.; Barsic, B.; Bouza, E.; Cecchi, E.; Moreno, A.; Doco-Lecompte, T.; Eisen, D.P.; Fortes, C.Q.; Fowler, V.G.; et al. The relationship between the initiation of antimicrobial therapy and the incidence of stroke in infective endocarditis: An analysis from the ICE Prospective Cohort Study (ICE-PCS). Am. Heart J. 2007, 154, 1086–1094. [Google Scholar] [CrossRef]
- Thuny, F.; Di Salvo, G.; Belliard, O.; Avierinos, J.F.; Pergola, V.; Rosenberg, V.; Casalta, J.P.; Gouvernet, J.; Derumeaux, G.; Iarussi, D.; et al. Risk of embolism and death in infective endocarditis: Prognostic value of echocardiography: A prospective multicenter study. Circulation 2005, 112, 69–75, Erratum in Circulation 2005, 112, e125. [Google Scholar] [CrossRef]
- Nunes, M.C.; Gelape, C.L.; Ferrari, T.C. Profile of infective endocarditis at a tertiary care center in Brazil during a seven-year period: Prognostic factors and in-hospital outcome. Int. J. Infect. Dis. 2010, 14, e394–e398. [Google Scholar] [CrossRef] [Green Version]
- Di Salvo, G.; Habib, G.; Pergola, V.; Avierinos, J.F.; Philip, E.; Casalta, J.P.; Vailloud, J.M.; Derumeaux, G.; Gouvernet, J.; Ambrosi, P.; et al. Echocardiography predicts embolic events in infective endocarditis. J. Am. Coll. Cardiol. 2001, 37, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Vilacosta, I.; Graupner, C.; San Román, J.A.; Sarriá, C.; Ronderos, R.; Fernández, C.; Mancini, L.; Sanz, O.; Sanmartín, J.V.; Stoermann, W. Risk of embolization after institution of antibiotic therapy for infective endocarditis. J. Am. Coll. Cardiol. 2002, 39, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Nappi, F.; Spadaccio, C.; Dreyfus, J.; Attias, D.; Acar, C.; Bando, K. Mitral endocarditis: A new management framework. J. Thorac. Cardiovasc. Surg. 2018, 156, 1486–1495.e4. [Google Scholar] [CrossRef] [Green Version]
- Avtaar Singh, S.S.; Costantino, M.F.; D’Addeo, G.; Cardinale, D.; Fiorilli, R.; Nappi, F. A narrative review of diagnosis of infective endocarditis-imaging methods and comparison. Ann. Transl. Med. 2020, 8, 1621. [Google Scholar] [CrossRef] [PubMed]
- Duval, X.; Iung, B.; Klein, I.; Thabut, G.; Arnoult, F.; Lepage, L.; Laissy, J.P.; Wolff, M.; Leport, C. IMAGE (Resonance Magnetic Imaging at the Acute Phase of Endocarditis) Study Group. Effect of early cerebral magnetic resonance imaging on clinical decisions in infective endocarditis: A prospective study. Ann. Intern. Med. 2010, 152, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Béraud, G.; Tubiana, S.; Erpelding, M.-L.; Le Moing, V.; Chirouze, C.; Gorenne, I.; Manchon, P.; Tattevin, P.; Vernet, V.; Varon, E.; et al. Combined Bacterial Meningitis and Infective Endocarditis: When Should We Search for the Other When Either One is Diagnosed? Infect. Dis. Ther. 2022, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Vitali, P.; Savoldi, F.; Segati, F.; Melazzini, L.; Zanardo, M.; Fedeli, M.P.; Benedek, A.; Di Leo, G.; Menicanti, L.; Sardanelli, F. MRI versus CT in the detection of brain lesions in patients with infective endocarditis before or after cardiac surgery. Neuroradiology 2021, 64, 905–913. [Google Scholar] [CrossRef]
- Corr, P.; Wright, M.; Handler, L.C. Endocarditis- related cerebral aneurysms: Radiologic changes with treatment. AJNR Am. J. Neuroradiol. 1995, 16, 745–748. [Google Scholar]
- Champey, J.; Pavese, P.; Bouvaist, H.; Maillet, M.; Kastler, A.; Boussat, B.; Francois, P.; The Investigator Groups. Is brain angio-MRI useful in infective endocarditis management? Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 2053–2058. [Google Scholar] [CrossRef]
- Peters, P.J.; Harrison, T.; Lennox, J.L. A dangerous dilemma: Management of infectious intracranial aneurysms complicating endocarditis. Lancet Infect. Dis 2006, 6, 742–748. [Google Scholar] [CrossRef]
- Serrano, F.; Guédon, A.; Saint-Maurice, J.-P.; Labeyrie, M.-A.; Civelli, V.; Eliezer, M.; Houdart, E. Endovascular treatment of infectious intracranial aneurysms complicating infective endocarditis: A series of 31 patients with 55 aneurysms. Neuroradiology 2021, 64, 353–360. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C.; Mihos, C. Infective endocarditis in the 21st century. Ann. Transl. Med. 2020, 8, 1620. [Google Scholar] [CrossRef]
- Slipczuk, L.; Codolosa, J.N.; Davila, C.D.; Romero-Corral, A.; Yun, J.; Pressman, G.S.; Figueredo, V.M. Infective Endocarditis Epidemiology Over Five Decades: A Systematic Review. PLoS ONE 2013, 8, e82665. [Google Scholar] [CrossRef]
- Voigt, A.; Shalaby, A.; Saba, S. Rising rates of cardiac rhythm management device infections in the United States: 1996 through 2003. J. Am. Coll. Cardiol. 2006, 48, 590–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nappi, F.; Iervolino, A.; Singh, S.S.A. The New Challenge for Heart Endocarditis: From Conventional Prosthesis to New Devices and Platforms for the Treatment of Structural Heart Disease. Biomed. Res. Int. 2021, 2021, 7302165. [Google Scholar] [CrossRef]
- Amat-Santos, I.J.; Messika-Zeitoun, D.; Eltchaninoff, H.; Kapadia, S.; Lerakis, S.; Cheema, A.N.; Gutiérrez-Ibañes, E.; Muñoz, A.; Pan, M.; Webb, J.G.; et al. Response to Letters Regarding Article, “Infective Endocarditis After Transcatheter Aortic Valve Implantation: Results From a Large Multicenter Registry”. Circulation 2015, 132, e372–e374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangner, N.; Woitek, F.; Haussig, S.; Schlotter, F.; Stachel, G.; Höllriegel, R.; Wilde, J.; Lindner, A.; Holzhey, D.; Leontyev, S.; et al. Incidence, Predictors, and Outcome of Patients Developing Infective Endocarditis Following Transfemoral Transcatheter Aortic Valve Replacement. J. Am. Coll. Cardiol. 2016, 67, 2907–2908. [Google Scholar] [CrossRef] [PubMed]
- Van Dijck, I.; Budts, W.; Cools, B.; Eyskens, B.; Boshoff, D.E.; Heying, R.; Frerich, S.; Vanagt, W.Y.; Troost, E.; Gewillig, M. Infective endocarditis of a transcatheter pulmonary valve in comparison with surgical implants. Heart 2015, 101, 788–793. [Google Scholar] [CrossRef] [Green Version]
- Duval, X.; Delahaye, F.; Alla, F.; Tattevin, P.; Obadia, J.F.; Le Moing, V.; Doco-Lecompte, T.; Celard, M.; Poyart, C.; Strady, C.; et al. The AEPEI Study Group;. Temporal trends in infective endocarditis in the context of prophylaxis guideline modifications: Three successive population-based surveys. J. Am. Coll. Cardiol. 2012, 59, 1968–1976. [Google Scholar] [CrossRef]
- Hoen, B.; Alla, F.; Selton-Suty, C.; Bouvet, A.; Briançon, S.; Casalta, J.P.; Danchin, N.; Delahaye, F.; Etienne, J.; Le Moing, V.; et al. Changing profile of infective endocarditis: Results of a 1-year survey in France. JAMA 2002, 288, 75–81. [Google Scholar] [CrossRef]
- Yew, H.S.; Murdoch, D.R. Global trends in infective endocarditis epidemiology. Curr. Infect. Dis. Rep. 2012, 14, 367–372. [Google Scholar] [CrossRef]
- Carapetis, J.R.; Steer, A.C.; Mulholland, E.K.; Weber, M. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 2005, 5, 685–694. [Google Scholar] [CrossRef]
- Marijon, E.; Ou, P.; Celermajer, D.S.; Ferreira, B.; Mocumbi, A.O.; Jani, D.; Paquet, C.; Jacob, S.; Sidi, D.; Jouven, X. Prevalence of Rheumatic Heart Disease Detected by Echocardiographic Screening. N. Engl. J. Med. 2007, 357, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Rwebembera, J.; Nascimento, B.R.; Minja, N.W.; de Loizaga, S.; Aliku, T.; dos Santos, L.P.A.; Galdino, B.F.; Corte, L.S.; Silva, V.R.; Chang, A.Y.; et al. Recent Advances in the Rheumatic Fever and Rheumatic Heart Disease Continuum. Pathogens 2022, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Seckeler, M.D.; Hoke, T.R. The worldwide epidemiology of acute rheumatic fever and rheumatic heart disease. Clin. Epidemiol. 2011, 3, 67–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belay, W.; Dessie, A.; Ahmed, H.; Gedlu, E.; Mariyo, A.; Shehibo, A.; Tigabu, Z.; Aliyu, M.H.; Soslow, J. Secondary prevention of rheumatic heart disease in Ethiopia: A multicenter study. BMC Cardiovasc. Disord. 2022, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Correa de Sa, D.D.C.; Tleyjeh, I.; Anavekar, N.S.; Schultz, J.C.; Thomas, J.M.; Lahr, B.D.; Bachuwar, A.; Pazdernik, M.; Steckelberg, J.M.; Wilson, W.R.; et al. Epidemiological Trends of Infective Endocarditis: A Population-Based Study in Olmsted County, Minnesota. Mayo Clin. Proc. 2010, 85, 422–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baddour, L.M.; Shafiyi, A.; Lahr, B.D.; Anavekar, N.S.; Steckelberg, J.M.; Wilson, W.R.; Sohail, M.R.; DeSimone, D.C. A Contemporary Population-Based Profile of Infective Endocarditis Using the Expanded Rochester Epidemiology Project. Mayo Clin. Proc. 2021, 96, 1438–1445. [Google Scholar] [CrossRef]
- Fowler, V.G., Jr.; Miro, J.M.; Hoen, B.; Cabell, C.H.; Abrutyn, E.; Rubinstein, E.; Corey, G.R.; Spelman, D.; Bradley, S.F.; Barsic, B.; et al. The ICE Investigators; Staphylococcus aureus endocarditis: A consequence of medical progress. JAMA 2005, 293, 3012–3021. [Google Scholar] [CrossRef] [Green Version]
- Marks, L.R.; Calix, J.J.; Wildenthal, J.A.; Wallace, M.A.; Sawhney, S.S.; Ransom, E.M.; Durkin, M.J.; Henderson, J.P.; Burnham, C.-A.D.; Dantas, G. Staphylococcus aureus injection drug use-associated bloodstream infections are propagated by community outbreaks of diverse lineages. Commun. Med. 2021, 1, 52. [Google Scholar] [CrossRef]
- Nappi F, Spadaccio C, Chello M, Acar C The Ross procedure: Underuse or under-comprehension? J Thorac Cardiovasc Surg. 2015, 149, 1463–1464. [CrossRef] [Green Version]
- Benedetto, U.; Avtaar Singh, S.S.; Spadaccio, C.; Moon, M.R.; Nappi, F. A narrative review of the interpretation of guidelines for the treatment of infective endocarditis. Ann. Transl. Med. 2020, 8, 1623. [Google Scholar] [CrossRef]
- Benedetto, U.; Spadaccio, C.; Gentile, F.; Moon, M.R.; Nappi, F. A narrative review of early surgery versus conventional treatment for infective endocarditis: Do we have an answer? Ann. Transl. Med. 2020, 8, 1626. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C.; Moon, M.R. A management framework for left sided endocarditis: A narrative review. Ann. Transl. Med. 2020, 8, 1627. [Google Scholar] [CrossRef] [PubMed]
- Pollari, F.; Ziegler, R.; Nappi, F.; Grossmann, I.; Steinmann, J.; Fischlein, T. Redo aortic valve replacement for prosthesis endocarditis in patients previously classified as high or prohibitive risk: A narrative review. Ann. Transl. Med. 2020, 8, 1629. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Singh, S.S.A.; Spadaccio, C.; Acar, C. Revisiting the guidelines and choice the ideal substitute for aortic valve endocarditis. Ann. Transl. Med. 2020, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Nenna, A.; Petitti, T.; Spadaccio, C.; Gambardella, I.; Lusini, M.; Chello, M.; Acar, C. Long-term outcome of cryopreserved allograft for aortic valve replacement. J. Thorac. Cardiovasc. Surg. 2018, 156, 1357–1365.e6. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Spadaccio, C.; Acar, C. Use of allogeneic tissue to treat infective valvular disease: Has everything been said? J. Thorac. Cardiovasc. Surg. 2017, 153, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Nappi, F.; Spadaccio, C. Keep fumbling around in the dark when it comes to infective endocarditis, or produce new, reliable data to redesign the guidelines? J. Thorac. Cardiovasc. Surg. 2018, 155, 75–76. [Google Scholar] [CrossRef] [Green Version]
- Lyytikainen, O.; Ruotsalainen, E.; Jarvinen, A.; Valtonen, V.; Ruutu, P. Trends and outcome of nosocomial and community-acquired bloodstream infections due to Staphylococcus aureus in Finland, 1995–2001. Eur. J. Clin. Microbiol. Infect. Dis. 2005, 24, 399–404. [Google Scholar] [CrossRef]
- Ammerlaan, H.S.M.; Harbarth, S.; Buiting, A.G.M.; Crook, D.W.; Fitzpatrick, F.; Hanberger, H.; Herwaldt, L.A.; Van Keulen, P.H.J.; Kluytmans, J.A.J.W.; Kola, A.; et al. Secular Trends in Nosocomial Bloodstream Infections: Antibiotic-Resistant Bacteria Increase the Total Burden of Infection. Clin. Infect. Dis. 2012, 56, 798–805. [Google Scholar] [CrossRef]
- Forsblom, E.; Kakriainen, A.; Ruotsalainen, E.; Järvinen, A. Methicillin-sensitive Staphylococcus aureus bacteremia in aged patients: The importance of formal infectious specialist consultation. Eur. Geriatr. Med. 2018, 9, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, I.; Millon, B.; Meugnier, H.; Vandenesch, F.; Maurin, M.; Pavese, P.; Boisset, S. High prevalence of spa type t571 among methicillin-susceptible Staphylococcus aureus from bacteremic patients in a French University Hospital. PLoS ONE 2018, 13, e0204977. [Google Scholar] [CrossRef] [Green Version]
- Greenspon, A.J.; Patel, J.D.; Lau, E.; Ochoa, J.A.; Frisch, D.R.; Ho, R.T.; Pavri, B.B.; Kurtz, S.M. 16-year trends in the infection burden for pacemakers and implantable cardioverter-defibrillator in the United States 1993 to 2008. J. Am. Coll. Cardiol. 2011, 58, 1001–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traykov, V.; Blomström-Lundqvist, C. Antibiotic-Eluting Envelopes for the Prevention of Cardiac Implantable Electronic Device Infections: Rationale, Efficacy, and Cost-Effectiveness. Front. Cardiovasc. Med. 2022, 9, 855233. [Google Scholar] [CrossRef] [PubMed]
- Elad, B.; Perl, L.; Hamdan, A.; Yahav, D.; Atamna, A.; Shaked, H.; Rubchevsky, V.; Sharony, R.; Bernstine, H.; Shapira, Y.; et al. The clinical value of the endocarditis team: Insights from before and after guidelines implementation strategy. Infection 2021, 50, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Han, H.C.; Hawkins, N.M.; Pearman, C.M.; Birnie, D.H.; Krahn, A.D. Epidemiology of cardiac implantable electronic device infections: Incidence and risk factors. Europace 2021, 23 (Suppl. 4), iv3–iv10. [Google Scholar] [CrossRef] [PubMed]
- Day, M.D.; Gauvreau, K.; Shulman, S.; Newburger, J.W. Characteristics of children hospitalized with infective endocarditis. Circulation 2009, 119, 865–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, A.L.; Tani, L.Y.; Weng, H.Y.; Wilkes, J.; Menon, S.C. Resource utilization and outcomes of infective endocarditis in children. J. Pediatr. 2014, 165, 807–812.e1. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, J.; Miller, S.; Shirley, D.A. Extended Interval Aminoglycoside Treatment for Klebsiella Pneumoniae Endocarditis in an Extremely Low Birth Weight Neonate. J. Pediatr. Pharmacol. Ther. 2022, 27, 85–89. [Google Scholar] [CrossRef]
- Rushani, D.; Kaufman, J.S.; Ionescu-Ittu, R.; Mackie, A.S.; Pilote, L.; Therrien, J.; Marelli, A.J. Infective endocarditis in children with congenital heart disease: Cumulative incidence and predictors. Circulation 2013, 128, 1412–1419. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.C.; Lai, C.C.; Wang, C.Y.; Wang, Y.H.; Wang, J.Y.; Hsu, Y.L.; Hu, Y.L.; Wu, E.T.; Lin, M.T.; Sy, L.B.; et al. Risk factors for infective endocarditis in children with congenital heart diseases—A nationwide population-based case control study. Int. J. Cardiol. 2017, 248, 126–130. [Google Scholar] [CrossRef]
- Snygg-Martin, U.; Giang, K.W.; Dellborg, M.; Robertson, J.; Mandalenakis, Z. Cumulative Incidence of Infective Endocarditis in Patients with Congenital Heart Disease: A Nationwide, Case-Control Study Over Nine Decades. Clin. Infect. Dis. 2021, 73, 1469–1475. [Google Scholar] [CrossRef]
- Morris, C.D.; Reller, M.D.; Menashe, V.D. Thirty-year incidence of infective endocarditis after surgery for congenital heart defect. JAMA 1998, 279, 599–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havers-Borgersen, E.; Butt, J.H.; Smerup, M.; Gislason, G.H.; Torp-Pedersen, C.; Gröning, M.; Schmidt, M.R.; Søndergaard, L.; Køber, L.; Fosbøl, E.L. Incidence of Infective Endocarditis Among Patients With Tetralogy of Fallot. J. Am. Heart Assoc. 2021, 10, e022445. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Spadaccio, C.; Mihos, C.; Shaikhrezai, K.; Acar, C.; Moon, M.R. The quest for the optimal surgical management of tricuspid valve endocarditis in the current era: A narrative review. Ann. Transl. Med. 2020, 8, 1628. [Google Scholar] [CrossRef]
- Valente, A.M.; Jain, R.; Scheurer, M.; Fowler, V.G.; Corey, G.R.; Bengur, A.R.; Sanders, S.; Li, J.S. Frequency of Infective Endocarditis Among Infants and Children With Staphylococcus aureus Bacteremia. Pediatrics 2005, 115, e15–e19. [Google Scholar] [CrossRef] [Green Version]
- Bendig, E.A.; Singh, J.; Butler, T.J.; Arrieta, A.C. The impact of the central venous catheter on the diagnosis of infectious endocarditis using Duke criteria in children with Staphylococcus aureus bacteremia. Pediatr. Infect. Dis. J. 2008, 27, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Kumarachandran, G.; Johnson, J.K.; Shirley, D.A.; Graffunder, E.; Heil, E.L. Predictors of Adverse Outcomes in Children With Staphylococcus aureus Bacteremia. J. Pediatr. Pharmacol. Ther. 2017, 22, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Durante-Mangoni, E.; Bradley, S.; Selton-Suty, C.; Tripodi, M.F.; Barsic, B.; Bouza, E.; Cabell, C.H.; Ramos, A.I.; Fowler, V., Jr.; Hoen, B.; et al. Results of the International Collaboration on Endocarditis Prospective Cohort Study. Arch. Intern. Med. 2008, 168, 2095–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampino, R.; Iossa, D.; Ursi, M.P.; Bertolino, L.; Karruli, A.; Molaro, R.; Esposito, G.; Vitrone, M.; D’Amico, F.; Albisinni, R.; et al. Clinical Significance and Prognostic Value of Hemostasis Parameters in 337 Patients with Acute Infective Endocarditis. J. Clin. Med. 2021, 10, 5386. [Google Scholar] [CrossRef] [PubMed]
- Molton, J.S.; Tambyah, P.A.; Ang, B.S.P.; Ling, M.L.; Fisher, D.A. The global spread of healthcare-associated multidrug-resistant bacteria: A perspective from Asia. Clin. Infect. Dis. 2013, 56, 1310–1318. [Google Scholar]
- Çaǧlayan, Ç.; Barnes, S.L.; Pineles, L.L.; Harris, A.D.; Klein, E.Y. A Data-Driven Framework for Identifying Intensive Care Unit Admissions Colonized With Multidrug-Resistant Organisms. Front. Public Health 2022, 10, 853757. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef] [Green Version]
- López, J.; Revilla, A.; Vilacosta, I.; Villacorta, E.; González-Juanatey, C.; Gómez, I.; Rollán, M.J.; Román, J.A.S. Definition, clinical profile, microbiological spectrum, and prognostic factors of early-onset prosthetic valve endocarditis. Eur. Hear. J. 2007, 28, 760–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Valle, H.; Fariñas-Álvarez, C.; García-Palomo, J.D.; Bernal, J.M.; Martín-Durán, R.; Díez, J.F.G.; Revuelta, J.M.; Fariñas, M.C. Clinical course and predictors of death in prosthetic valve endocarditis over a 20-year period. J. Thorac. Cardiovasc. Surg. 2010, 139, 887–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Chen, L.; Chen, X.; Tang, A.; Huang, D.; Pan, Q.; Fang, Z. Prevalence and Molecular Characterization of Methicillin-Resistant Staphylococci Recovered from Public Shared Bicycles in China. Int. J. Environ. Res. Public Health 2022, 19, 4492. [Google Scholar] [CrossRef] [PubMed]
- Argemi, X.; Hansmann, Y.; Prola, K.; Prévost, G. Coagulase-Negative Staphylococci Pathogenomics. Int. J. Mol. Sci. 2019, 20, 1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, V.H.; Woods, C.W.; Miro, J.M.; Hoen, B.; Cabell, C.H.; Pappas, P.A.; Federspiel, J.; Athan, E.; Stryjewski, M.E.; Nacinovich, F.; et al. The International Collaboration on Endocarditis-Prospective Cohort Study Group.Emergence of coagulase-negative staphylococci as a cause of native valve endocarditis. Clin. Infect. Dis. 2008, 46, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Chu, V.H.; Miro, J.M.; Hoen, B.; Cabell, C.H.; Pappas, P.A.; Jones, P.; Stryjewski, M.; Anguera, I.; Braun, S.; Muñoz, P.; et al. Coagulase-negative staphylococcal prosthetic valve endocarditis--a contemporary update based on the International Collaboration on Endocarditis: Prospective cohort study. Heart 2008, 95, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Babeș, E.E.; Lucuța, D.A.; Petcheși, C.D.; Zaha, A.A.; Ilyes, C.; Jurca, A.D.; Vesa, C.M.; Zaha, D.C.; Babeș, V.V. Clinical Features and Outcome of Infective Endocarditis in a University Hospital in Romania. Medicina 2021, 57, 158. [Google Scholar] [CrossRef] [PubMed]
- Nigo, M.; Munita, J.M.; Arias, C.A.; Murray, B.E. What’s new in the treatment of enterococcal endocarditis? Curr. Infect. Dis. Rep. 2014, 16, 431. [Google Scholar] [CrossRef] [Green Version]
- Brouqui, P.; Raoult, D. Endocarditis due to rare and fastidious bacteria. Clin. Microbiol. Rev. 2001, 14, 177–207. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Badley, A.D.; Cockerill, F.R.; Steckelberg, J.M.; Wilson, W.R. Infective endocarditis caused by HACEK microorganisms. Annu. Rev. Med. 1997, 48, 25–33. [Google Scholar] [CrossRef]
- Oumarou Hama, H.; Aboudharam, G.; Barbieri, R.; Lepidi, H.; Drancourt, M. Immunohistochemical diagnosis of human infectious diseases: A review. Diagn. Pathol. 2022, 17, 17. [Google Scholar] [CrossRef]
- Baddley, J.W.; Benjamin, D.K., Jr.; Patel, M.; Miró, J.; Athan, E.; Barsic, B.; Bouza, E.; Clara, L.; Elliott, T.; Kanafani, Z.; et al. The International Collaboration on Endocarditis-Prospective Cohort Study Group (ICE-PCS).Candida infective endocarditis. Eur. J. Clin. Microbiol. Infect. Dis. 2008, 27, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Huggins, J.P.; Hohmann, S.; David, M.Z. Candida Infective Endocarditis: A Retrospective Study of Patient Characteristics and Risk Factors for Death in 703 United States Cases, 2015–2019. Open Forum. Infect Dis. 2020, 8, ofaa628. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, F.A.; Christophersen, L.; Laulund, A.S.; Lundquist, R.; Lerche, C.; Nielsen, P.R.; Bundgaard, H.; Høiby, N.; Moser, C. Novel human in vitro vegetation simulation model for infective endocarditis. APMIS 2021, 129, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, E.G.; Rimoldi, S.G.; Cavallo, I.; D’Agosto, G.; Trento, E.; Cagnoni, G.; Palazzin, A.; Pagani, C.; Romeri, F.; De Vecchi, E.; et al. Microbial biofilm correlates with an increased antibiotic tolerance and poor therapeutic outcome in infective endocarditis. BMC Microbiol. 2019, 19, 228. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, F.A.; Nielsen, L.; Struve Andersen, J.; Bock, M.; Christophersen, L.; Sunnerhagen, T.; Lerche, C.J.; Bay, L.; Bundgaard, H.; Høiby, N.; et al. Dynamics of a Staphylococcus aureus infective endocarditis simulation model. APMIS 2022, 130, 515–523. [Google Scholar] [CrossRef] [PubMed]
- McAdow, M.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus secretes coagulase and von Willebrand factor binding protein to modify the coagulation cascade and establish host infections. J. Innate Immun. 2012, 4, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomer, L.; Schneewind, O.; Missiakas, D. Multiple ligands of von Willebrand factor-binding protein (vWbp) promote Staphylococcus aureus clot formation in human plasma. J. Biol. Chem. 2013, 288, 28283–28292. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.P.; Kang, M.; Ganesh, V.K.; Ravirajan, D.; Li, B.; Höök, M. Coagulase and Efb of Staphylococcus aureus Have a Common Fibrinogen Binding Motif. mBio 2016, 7, e01885-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Liu, W.; Arora, S.; Ganesh, V.; Ko, Y.P.; Höök, M. The Complex Fibrinogen Interactions of the Staphylococcus aureus Coagulases. Front. Cell. Infect. Microbiol. 2019, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Arora, S.; Liu, W.; Churion, K.; Wu, Y.; Höök, M. vhp Is a Fibrinogen-Binding Protein Related to vWbp in Staphylococcus aureus. mBio 2021, 12, e0116721. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.; Töre, Y.; Hoesker, V.; Ameling, S.; Grün, K.; Völker, U.; Schulze, P.C.; Franz, M.; Faber, C.; Schaumburg, F.; et al. Host-pathogen interactions of clinical S. aureus isolates to induce infective endocarditis. Virulence 2021, 12, 2073–2087. [Google Scholar] [CrossRef]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia associated with toothbrushing and dental extraction. Circulation 2008, 117, 3118–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmer, E.; Que, Y.A.; Entenza, J.M.; Moreillon, P. New concepts in the pathophysiology of infective endocarditis. Curr. Infect. Dis. Rep. 2006, 8, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreillon, P.; Que, Y.A.; Bayer, A.S. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect. Dis. Clin. N. Am. 2002, 16, 297–318. [Google Scholar] [CrossRef]
- Mancini, S.; Oechslin, F.; Menzi, C.; Que, Y.A.; Claes, J.; Heying, R.; Veloso, T.R.; Vanassche, T.; Missiakas, D.; Schneewind, O.; et al. Marginal role of von Willebrand factor-binding protein and coagulase in the initiation of endocarditis in rats with catheter-induced aortic vegetations. Virulence 2018, 9, 1615–1624. [Google Scholar] [CrossRef] [Green Version]
- Werdan, K.; Dietz, S.; Löffler, B.; Niemann, S.; Bushnaq, H.; Silber, R.-E.; Peters, G.; Müller-Werdan, U. Mechanisms of infective endocarditis: Pathogen–host interaction and risk states. Nat. Rev. Cardiol. 2013, 11, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, A.; Linke, A.; Latib, A.; Ihlemann, N.; Urena, M.; Walther, T.; Husser, O.; Herrmann, C.; Nombela-Franco, L.; Cheema, A.; et al. Infective Endocarditis Following Transcatheter Aortic Valve Replacement: Comparison of Balloon- Versus Self-Expandable Valves. Circ. Cardiovasc. Interv. 2019, 12, e007938. [Google Scholar] [CrossRef]
- Rodríguez-Vidigal, F.F.; Nogales-Asensio, J.M.; Calvo-Cano, A.; González-Fernández, R.; Martínez-Carapeto, A.; Gómez-Sanchez, I.; Bengla-Limpo, B.; Merchán-Herrera, A.; Nogales-Muñoz, N.; Vera-Tomé, A.; et al. Infective endocarditis after transcatheter aortic valve implantation: Contributions of a single-centre experience on incidence and associated factors. Enfermedades Infecc. y Microbiol. Clin. (English ed.) 2019, 37, 428–434. [Google Scholar] [CrossRef]
- Ciofu, O.; Moser, C.; Jensen, P.Ø.; Høiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Annappah, D.; Saling, M.; Prodafikas, J.; Badie, A.N. Device-associated aortic valve endocarditis due to a complicated Enterobacter cloacae urinary tract infection. IDCases 2021, 27, e01365. [Google Scholar] [CrossRef]
- Di Carluccio, C.; Forgione, R.E.R.; Bosso, A.; Yokoyama, S.; Manabe, Y.; Pizzo, E.; Molinaro, A.; Fukase, K.; Fragai, M.; Bensing, B.A.; et al. Molecular recognition of sialoglycans by streptococcal Siglec-like adhesins: Toward the shape of specific inhibitors. RSC Chem. Biol. 2021, 2, 1618–1630. [Google Scholar] [CrossRef]
- Manukumar, H.M.; Umesha, S. MALDI-TOF-MS based identification and molecular characterization of food associated methicillin-resistant Staphylococcus aureus. Sci. Rep. 2017, 7, 11414. [Google Scholar] [CrossRef]
- Mempel, M.; Schnopp, C.; Hojka, M.; Fesq, H.; Weidinger, S.; Schaller, M.; Korting, H.; Ring, J.; Abeck, D. Invasion of human keratinocytes by Staphylococcus aureus and intracellular bacterial persistence represent haemolysin-independent virulence mechanisms that are followed by features of necrotic and apoptotic keratinocyte cell death. Br. J. Dermatol. 2002, 146, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Matsumoto, M.; Katayama, Y.; Oguma, R.; Wakabayashi, S.; Nygaard, T.; Saijo, S.; Inohara, N.; Otto, M.; Matsue, H.; et al. Staphylococcus aureus Virulent PSMα Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe 2017, 22, 667–677.e5. [Google Scholar] [CrossRef] [Green Version]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef] [Green Version]
- Kawa, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Kupper, T.S.; Fuhlbrigge, R.C. Immune surveillance in the skin: Mechanisms and clinical consequences. Nat. Rev. Immunol. 2004, 4, 211–222. [Google Scholar] [CrossRef]
- Nestle, F.O.; Di, M.P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malachowa, N.; Whitney, A.R.; Kobayashi, S.D.; Sturdevant, D.E.; Kennedy, A.D.; Braughton, K.R.; Shabb, D.W.; Diep, B.A.; Chambers, H.F.; Otto, M.; et al. Global Changes in Staphylococcus aureus Gene Expression in Human Blood. PLoS ONE 2011, 6, e18617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonzo, F., 3rd; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonzo, F., 3rd; Torres, V.J. Bacterial survival amidst an immune onslaught: The contribution of the Staphylococcus aureus leukotoxins. PLoS Pathog. 2013, 9, e1003143. [Google Scholar] [CrossRef] [Green Version]
- Cheung, G.Y.C.; Joo, H.-S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins—Critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005, 3, 48–58. [Google Scholar] [CrossRef]
- Silverman, G.J.; Goodyear, C.S. Confounding B-cell defences: Lessons from a staphylococcal superantigen. Nat. Rev. Immunol. 2006, 6, 465–475. [Google Scholar] [CrossRef]
- Kim, H.K.; Cheng, A.G.; Kim, H.Y.; Missiakas, D.M.; Schneewind, O. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J. Exp. Med. 2010, 207, 1863–1870. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Frankel, M.B.; Schneewind, O.; Missiakas, D. Release of protein A from the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2014, 111, 1574–1579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Marichannegowda, M.H.; Rakesh, K.P.; Qin, H.L. Master mechanisms of Staphylococcus aureus: Consider its excellent protective mechanisms hindering vaccine development! Microbiol. Res. 2018, 212–213, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Z.; Hua, Y.H.; Yu, B.; Lau, C.C.Y.; Cai, J.P.; Zheng, S.Y.; Yam, W.C.; Kao, R.Y.T.; Sze, K.H.; Zheng, B.J.; et al. Recombinant ESAT-6-Like Proteins Provoke Protective Immune Responses against Invasive Staphylococcus aureus Disease in a Murine Model. Infect. Immun. 2015, 83, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, R.A.; Mocca, C.P.; Prabhakara, R.; Plaut, R.; Shirtliff, M.; Merkel, T.J.; Burns, D.L. Evaluation of Genetically Inactivated Alpha Toxin for Protection in Multiple Mouse Models of Staphylococcus aureus Infection. PLoS ONE 2013, 8, e63040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Ledue, O.; Jun, M.; Goulart, C.; Malley, R.; Lu, Y.J. Protection against Staphylococcus aureus Colonization and Infection by B- and T-Cell-Mediated Mechanisms. mBio 2018, 9, e01949-18. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Yao, D.; Yu, S.; Wang, X.; Li, X.; Wang, M.; Liu, S.; Feng, Z.; Chen, X.; Li, W.; et al. Protective humoral and CD4+ T cellular immune responses of Staphylococcus aureus vaccine MntC in a murine peritonitis model. Sci. Rep. 2018, 8, 3580. [Google Scholar] [CrossRef]
- Que, Y.-A.; Haefliger, J.-A.; Piroth, L.; Francois, P.; Widmer, E.; Entenza, J.M.; Sinha, B.N.M.; Herrmann, M.; Francioli, P.; Vaudaux, P.; et al. Fibrinogen and fibronectin binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J. Exp. Med. 2005, 201, 1627–1635. [Google Scholar] [CrossRef]
- Edwards, A.M.; Bowden, M.G.; Brown, E.L.; Laabei, M.; Massey, R.C. Staphylococcus aureus extracellular adherence protein triggers TNFα release, promoting attachment to endothelial cells via protein A. PLoS ONE 2012, 7, e43046. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The interaction of bacterial pathogens with platelets. Nat. Rev. Microbiol. 2006, 4, 445–457. [Google Scholar] [CrossRef]
- Veloso, T.R.; Chaouch, A.; Roger, T.; Giddey, M.; Vouillamoz, J.; Majcherczyk, P.; Que, Y.-A.; Rousson, V.; Moreillon, P.; Entenza, J.M. Use of a Human-Like Low-Grade Bacteremia Model of Experimental Endocarditis To Study the Role of Staphylococcus aureus Adhesins and Platelet Aggregation in Early Endocarditis. Infect. Immun. 2013, 81, 697–703. [Google Scholar] [CrossRef] [Green Version]
- Sinha, B.; Herrmann, M. Mechanism and consequences of invasion of endothelial cells by Staphylococcus aureus. Thromb. Haemost. 2005, 94, 266–277. [Google Scholar] [CrossRef]
- Hussain, M.; Haggar, A.; Heilmann, C.; Peters, G.; Flock, J.-I.; Herrmann, M. Insertional Inactivation of eap in Staphylococcus aureus Strain Newman Confers Reduced Staphylococcal Binding to Fibroblasts. Infect. Immun. 2002, 70, 2933–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palankar, R.; Binsker, U.; Haracska, B.; Wesche, J.; Greinacher, A.; Hammerschmidt, S. Interaction between the Staphylococcus aureus extracellular adherence protein Eap and its subdomains with platelets. Int. J. Med Microbiol. 2018, 308, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Haggar, A.; Peters, G.; Chhatwal, G.S.; Herrmann, M.; Flock, J.I.; Sinha, B. More than one tandem repeat domain of the extracellular adherence protein of Staphylococcus aureus is required for aggregation, adherence, and host cell invasion but not for leukocyte activation. Infect. Immun. 2008, 76, 5615–5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harraghy, N.; Hussain, M.; Haggar, A.; Chavakis, T.; Sinha, B.; Herrmann, M.; Flock, J.-I. The adhesive and immunomodulating properties of the multifunctional Staphylococcus aureus protein Eap. Microbiology 2003, 149, 2701–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heying, R.; van de Gevel, J.; Que, Y.A.; Moreillon, P.; Beekhuizen, H. Fibronectin-binding proteins and clumping factor A in Staphylococcus aureus experimental endocarditis: FnBPA is sufficient to activate human endothelial cells. Thromb. Haemost. 2007, 97, 617–626. [Google Scholar] [CrossRef]
- Piroth, L.; Que, Y.-A.; Widmer, E.; Panchaud, A.; Piu, S.; Entenza, J.M.; Moreillon, P. The Fibrinogen- and Fibronectin-Binding Domains of Staphylococcus aureus Fibronectin-Binding Protein A Synergistically Promote Endothelial Invasion and Experimental Endocarditis. Infect. Immun. 2008, 76, 3824–3831. [Google Scholar] [CrossRef] [Green Version]
- Claes, J.; Vanassche, T.; Peetermans, M.; Liesenborghs, L.; Vandenbriele, C.; Vanhoorelbeke, K.; Missiakas, D.; Schneewind, O.; Hoylaerts, M.F.; Heying, R.; et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor–binding protein. Blood 2014, 124, 1669–1676. [Google Scholar] [CrossRef]
- Pappelbaum, K.I.; Gorzelanny, C.; Grässle, S.; Suckau, J.; Laschke, M.W.; Bischoff, M.; Bauer, C.; Schorpp-Kistner, M.; Weidenmaier, C.; Schneppenheim, R.; et al. Ultralarge von Willebrand Factor Fibers Mediate Luminal Staphylococcus aureus Adhesion to an Intact Endothelial Cell Layer Under Shear Stress. Circulation 2013, 128, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Claes, J.; Liesenborghs, L.; Peetermans, M.; Veloso, T.R.; Missiakas, D.; Schneewind, O.; Mancini, S.; Entenza, J.M.; Hoylaerts, M.F.; Heying, R.; et al. Clumping factor A, von Willebrand factor-binding protein and von Willebrand factor anchor Staphylococcus aureus to the vessel wall. J. Thromb. Haemost. 2017, 15, 1009–1019. [Google Scholar] [CrossRef] [Green Version]
- Claes, J.; Ditkowski, B.; Liesenborghs, L.; Veloso, T.R.; Entenza, J.M.; Moreillon, P.; Vanassche, T.; Verhamme, P.; Hoylaerts, M.F.; Heying, R. Assessment of the Dual Role of Clumping Factor A in S. Aureus Adhesion to Endothelium in Absence and Presence of Plasma. Thromb. Haemost. 2018, 118, 1230–1241. [Google Scholar] [CrossRef] [Green Version]
- Na, M.; Hu, Z.; Mohammad, M.; Stroparo, M.D.N.; Ali, A.; Fei, Y.; Jarneborn, A.; Verhamme, P.; Schneewind, O.; Missiakas, D.; et al. The Expression of von Willebrand Factor-Binding Protein Determines Joint-Invading Capacity of Staphylococcus aureus, a Core Mechanism of Septic Arthritis. mBio 2020, 11, e02472-20. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. The remarkably multifunctional fibronectin binding proteins of Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1923–1931. [Google Scholar] [CrossRef]
- Ahmed, S.; Meghji, S.; Williams, R.J.; Henderson, B.; Brock, J.H.; Nair, S.P. Staphylococcus aureus fibronectin binding proteins are essential for internalization by osteoblasts but do not account for differences in intracellular levels of bacteria. Infect. Immun. 2001, 69, 2872–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massey, R.C.; Kantzanou, M.N.; Fowler, T.; Day, N.P.J.; Schofield, K.; Wann, E.R.; Berendt, A.R.; Hook, M.; Peacock, S.J. Fibronectin-binding protein A of Staphylococcus aureus has multiple, substituting, binding regions that mediate adherence to fibronectin and invasion of endothelial cells. Cell. Microbiol. 2001, 3, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.A.; Douglas, I.; Whawell, S.A. Differential adhesion and invasion by Staphylococcus aureus of epithelial cells derived from different anatomical sites. J. Med. Microbiol. 2012, 61 Pt 12, 1654–1661. [Google Scholar] [CrossRef] [Green Version]
- Niemann, S.; Nguyen, M.-T.; Eble, J.A.; Chasan, A.I.; Mrakovcic, M.; Böttcher, R.T.; Preissner, K.T.; Roßlenbroich, S.; Peters, G.; Herrmann, M. More Is Not Always Better—the Double-Headed Role of Fibronectin in Staphylococcus aureus Host Cell Invasion. mBio 2021, 12, e0106221. [Google Scholar] [CrossRef]
- Pietrocola, G.; Pellegrini, A.; Alfeo, M.J.; Marchese, L.; Foster, T.J.; Speziale, P. The iron-regulated surface determinant B (IsdB) protein from Staphylococcus aureus acts as a receptor for the host protein vitronectin. J. Biol. Chem. 2020, 295, 10008–10022. [Google Scholar] [CrossRef]
- Alfeo, M.J.; Pagotto, A.; Barbieri, G.; Foster, T.J.; Vanhoorelbeke, K.; De Filippis, V.; Speziale, P.; Pietrocola, G. Staphylococcus aureus iron-regulated surface determinant B (IsdB) protein interacts with von Willebrand factor and promotes adherence to endothelial cells. Sci. Rep. 2021, 11, 22799. [Google Scholar] [CrossRef]
- Leeten, K.; Jacques, N.; Lancellotti, P.; Oury, C. Aspirin or Ticagrelor in Staphylococcus aureus Infective Endocarditis: Where Do We Stand? Front. Cell. Dev. Biol. 2021, 9, 716302. [Google Scholar] [CrossRef]
- Ditkowski, B.; Bezulska-Ditkowska, M.; Jashari, R.; Baatsen, P.; Moreillon, P.; Rega, F.; Veloso, T.R.; Hoylaerts, M.F.; Heying, R.; Congenital Cardiology and Cardiac Surgery Group. Antiplatelet therapy abrogates platelet-assisted Staphylococcus aureus infectivity of biological heart valve conduits. J. Thorac. Cardiovasc. Surg. 2021, 161, e457–e472. [Google Scholar] [CrossRef]
- Hannachi, N.; Habib, G.; Camoin-Jau, L. Aspirin Effect on Staphylococcus aureus-Platelet Interactions During Infectious Endocarditis. Front. Med. 2019, 6, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Na, H.S.; Song, Y.R.; Shin, S.Y.; Kim, Y.M.; Chung, J. Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infect. Immun. 2014, 82, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, E.; Oho, T. Invasive Streptococcus mutans induces inflammatory cytokine production in human aortic endothelial cells via regulation of intracellular toll-like receptor 2 and nucleotide-binding oligomerization domain. Mol. Oral Microbiol. 2017, 32, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, K.; Na, H.; Jeong, S.; Park, H.R.; Kim, S.; Chung, J. Tumor necrosis factor-α and interleukin-1β expression pathway induced by Streptococcus mutans in macrophage cell line RAW 264.7. Mol. Oral Microbiol. 2012, 27, 149–159. [Google Scholar] [CrossRef]
- Tornos, P.; Gonzalez-Alujas, T.; Thuny, F.; Habib, G. Infective endocarditis: The European viewpoint. Curr. Probl. Cardiol. 2011, 36, 175–222. [Google Scholar] [CrossRef]
- Lockhart, P.B.; Brennan, M.T.; Thornhill, M.; Michalowicz, B.S.; Noll, J.; Bahrani-Mougeot, F.K.; Sasser, H.C. Poor oral hygiene as a risk factor for infective endocarditis-related bacteremia. J. Am. Dent. Assoc. 2009, 140, 1238–1244. [Google Scholar] [CrossRef]
- Lockhart, P.B.; Brennan, M.T.; Kent, M.L.; Norton, H.J.; Weinrib, D.A. Impact of amoxicillin prophylaxis on the incidence, nature, and duration of bacteremia in children after intubation and dental procedures. Circulation 2004, 109, 2878–2884. [Google Scholar] [CrossRef] [Green Version]
- Karikoski, E.; Junttila, K.; Järvinen, M.; Sarkola, T.; Blomqvist, M. Parental perceptions and experiences of an oral health care promotion intervention for children with congenital heart defects. Int. J. Qual. Stud. Health Well-Being 2022, 17, 2070968. [Google Scholar] [CrossRef]
- Okano, T.; Ashida, H.; Suzuki, S.; Shoji, M.; Nakayama, K.; Suzuki, T. Porphyromonas gingivalis triggers NLRP3-mediated inflammasome activation in macrophages in a bacterial gingipains-independent manner. Eur. J. Immunol. 2018, 48, 1965–1974. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Na, H.S.; Park, E.; Park, M.H.; Lee, H.A.; Chung, J. Streptococcus mutans activates the AIM2, NLRP3 and NLRC4 inflammasomes in human THP-1 macrophages. Int. J. Oral Sci. 2018, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.A.; Park, M.H.; Song, Y.; Na, H.S.; Chung, J. Role of Aggregatibacter actinomycetemcomitans-induced autophagy in inflammatory response. J. Periodontol. 2020, 91, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Uslan, D.Z.; Sohail, M.R.; St Sauver, J.L.; Friedman, P.A.; Hayes, D.L.; Stoner, S.M.; Wilson, W.R.; Steckelberg, J.M.; Baddour, L.M. Permanent pacemaker and implantable cardioverter defibrillator infection: A population based study. Arch. Intern. Med. 2007, 167, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, J.B.; Jørgensen, O.D.; Møller, M.; Arnsbo, P.; Mortensen, P.T.; Nielsen, J.C. Infection after pacemaker implantation: Infection rates and risk factors associated with infection in a population-based cohort study of 46299 consecutive patients. Eur. Heart J. 2011, 32, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Landolina, M.; Gasparini, M.; Lunati, M.; Iacopino, S.; Boriani, G.; Bonanno, C.; Vado, A.; Proclemer, A.; Capucci, A.; Zucchiatti, C.; et al. Cardiovascular Centers Participating in the ClinicalService Project.Long-term complications related to biventricular defibrillator implantation: Rate of surgical revisions and impact on survival: Insights from the Italian Clinical Service Database. Circulation 2011, 123, 2526–2535. [Google Scholar] [CrossRef]
- Sohail, M.R.; Henrikson, C.A.; Braid-Forbes, M.J.; Forbes, K.; Lerner, D.J. Increased Long-Term Mortality in Patients with Cardiovascular Implantable Electronic Device Infections. Pacing Clin. Electrophysiol. 2014, 38, 231–239. [Google Scholar] [CrossRef]
- Sohail, M.R.; Henrikson, C.A.; Braid-Forbes, M.J.; Forbes, K.F.; Lerner, D.J. Mortality and cost associated with cardiovascular implantable electronic device infections. Arch. Intern. Med. 2011, 171, 1821–1828. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, K.A.; Konstantelias, A.A.; Falagas, M.E. Risk factors for cardiac implantable electronic device infection: A systematic review and metaanalysis. Europace 2015, 17, 767–777. [Google Scholar] [CrossRef]
- Da Costa, A.; Kirkorian, G.; Cucherat, M.; Delahaye, F.; Chevalier, P.; Cerisier, A.; Isaaz, K.; Touboul, P. Antibiotic prophylaxis for permanent pacemaker implantation: A meta-analysis. Circulation 1998, 97, 1796–1801. [Google Scholar] [CrossRef]
- Blomstrom-Lundqvist, C.; Ostrowska, B. Prevention of cardiac implantable electronic device infections: Guidelines and conventional prophylaxis. Europace 2021, 23 (Suppl. 4), iv11–iv19. [Google Scholar] [CrossRef]
- de Oliveira, J.C.; Martinelli, M.; Nishioka, S.A.; Varejão, T.; Uipe, D.; Pedrosa, A.A.; Costa, R.; D’Avila, A.; Danik, S.B. Efficacy of antibiotic prophylaxis before the implantation of pacemakers and cardioverter-defibrillator: Results of a large, prospective, randomized, double-blinded, placebo-controlled trial. Circ. Arrhythm. Electrophysiol. 2009, 2, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Satriano, U.M.; Nenna, A.; Spadaccio, C.; Pollari, F.; Fischlein, T.; Chello, M.; Nappi, F. Guidelines on prosthetic heart valve management in infective endocarditis: A narrative review comparing American Heart Association/American College of Cardiology and European Society of Cardiology guidelines. Ann. Transl. Med. 2020, 8, 1625. [Google Scholar] [CrossRef] [PubMed]
- Pollari, F.; Spadaccio, C.; Cuomo, M.; Chello, M.; Nenna, A.; Fischlein, T.; Nappi, F. Sharing of decision-making for infective endocarditis surgery: A narrative review of clinical and ethical implications. Ann. Transl. Med. 2020, 8, 1624. [Google Scholar] [CrossRef] [PubMed]
- Mihos, C.G.; Nappi, F. A narrative review of echocardiography in infective endocarditis of the right heart. Ann. Transl. Med. 2020, 8, 1622. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Hidalgo, N.; Almirante, B.; Gavaldà, J.; Gurgui, M.; Peña, C.; de Alarcón, A.; Ruiz, J.; Vilacosta, I.; Montejo, M.; Vallejo, N.; et al. Ampicillin Plus Ceftriaxone Is as Effective as Ampicillin Plus Gentamicin for Treating Enterococcus faecalis Infective Endocarditis. Clin. Infect. Dis. 2013, 56, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Gavaldà, J.; Len, O.; Miró, J.M.; Muñoz, P.; Montejo, M.; Alarcón, A.; de la Torre-Cisneros, J.; Peña, C.; Martínez-Lacasa, X.; Sarria, C.; et al. Brief communication: Treatment of Enterococcus faecalis endocarditis with ampicillin plus ceftriaxone. Ann. Intern. Med. 2007, 146, 574–579. [Google Scholar] [CrossRef]
- Vahanian, A.; Beyersdorf, F.; Praz, F.; Milojevic, M.; Baldus, S.; Bauersachs, J.; Capodanno, D.; Conradi, L.; De Bonis, M.; De Paulis, R.; et al. ESC/EACTS Scientific Document Group2021 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J. 2022, 43, 561–632. [Google Scholar] [CrossRef] [PubMed]
- Victor, F.; De Place, C.; Camus, C.; Le Breton, H.; Leclercq, C.; Pavin, D.; Mabo, P.; Daubert, C. Pacemaker lead infection: Echocardiographic features, management, and outcome. Heart 1999, 81, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downey, B.C.; Juselius, W.E.; Pandian, N.G.; Estes, N.A.M.; Link, M.S. Incidence and Significance of Pacemaker and Implantable Cardioverter-Defibrillator Lead Masses Discovered during Transesophageal Echocardiography. Pacing Clin. Electrophysiol. 2011, 34, 679–683. [Google Scholar] [CrossRef]
- Sarrazin, J.-F.; Philippon, F.; Tessier, M.; Guimond, J.; Molin, F.; Champagne, J.; Nault, I.; Blier, L.; Nadeau, M.; Charbonneau, L.; et al. Usefulness of Fluorine-18 Positron Emission Tomography/Computed Tomography for Identification of Cardiovascular Implantable Electronic Device Infections. J. Am. Coll. Cardiol. 2012, 59, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F.Z.; James, J.; Cunnington, C.; Motwani, M.; Fullwood, C.; Hooper, J.; Burns, P.; Qamruddin, A.; Al-Bahrani, G.; Armstrong, I.; et al. Early diagnosis of cardiac implantable electronic device generator pocket infection using 18F-FDGPET/ CT. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Cautela, J.; Alessandrini, S.; Cammilleri, S.; Giorgi, R.; Richet, H.; Casalta, J.-P.; Habib, G.; Raoult, D.; Mundler, O.; Deharo, J.-C. Diagnostic yield of FDG positron-emission tomography/ computed tomography in patients with CEID infection: A pilot study. Europace 2013, 15, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoe, J.A.T.; Barlow, G.; Chambers, J.B.; Gammage, M.; Guleri, A.; Howard, P.; Olson, E.; Perry, J.D.; Prendergast, B.D.; Spry, M.J.; et al. Guidelines for the diagnosis, prevention and management of implantable cardiac electronic device infection. Report of a joint Working Party project on behalf of the British Society for Antimicrobial Chemotherapy (BSAC, host organization), British Heart Rhythm Society (BHRS), British Cardiovascular Society (BCS), British Heart Valve Society (BHVS) and British Society for Echocardiography (BSE). J. Antimicrob. Chemother. 2014, 70, 325–359. [Google Scholar] [PubMed]
- Baddour, L.M.; Epstein, A.E.; Erickson, C.C.; Knight, B.P.; Levison, M.E.; Lockhart, P.B.; Masoudi, F.A.; Okum, E.J.; Wilson, W.R.; Beerman, L.B.; et al. Council on Cardiovascular Surgery and Anesthesia, Council on Cardiovascular Nursing, Council on Clinical Cardiology, Interdisciplinary Council on Quality of Care and Outcomes Research. Update on cardiovascular implantable electronic device infections and their management: A scientific statement from the American Heart Association. Circulation 2010, 121, 458–477. [Google Scholar]
- Fu, H.X.; Huang, X.M.; Zhong, L.I.; Osborn, M.J.; Asirvatham, S.J.; Espinosa, R.E.; Brady, P.A.; Lee, H.C.; Greason, K.L.; Baddour, L.M.; et al. Outcomes and complications of lead removal: Can we establish a risk stratification schema for a collaborative and effective approach ? Pacing Clin. Electrophysiol. 2015, 38, 1439–1447. [Google Scholar] [CrossRef]
- Yuan, S.M. Mycobacterial endocarditis: A comprehensive review. Braz. J. Cardiovasc. Surg. 2015, 30, 3–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, N.; Monticelli, J.; Antonello, R.M.; Luzzati, R.; Gabrielli, M.; Ferrarese, M.; Codecasa, L.; Di Bella, S.; Giacobbe, D.R. Mycobacterium chimaera infections: An update. J. Infect. Chemother. 2020, 26, 199–205. [Google Scholar] [CrossRef]
- Estellés, A.; Woischnig, A.-K.; Liu, K.; Stephenson, R.; Lomongsod, E.; Nguyen, D.; Zhang, J.; Heidecker, M.; Yang, Y.; Simon, R.J.; et al. A High-Affinity Native Human Antibody Disrupts Biofilm from Staphylococcus aureus Bacteria and Potentiates Antibiotic Efficacy in a Mouse Implant Infection Model. Antimicrob. Agents Chemother. 2016, 60, 2292–2301. [Google Scholar] [CrossRef] [Green Version]
- Scialla, S.; Martuscelli, G.; Nappi, F.; Singh, S.S.A.; Iervolino, A.; Larobina, D.; Ambrosio, L.; Raucci, M.G. Trends in Managing Cardiac and Orthopedic Device-Associated Infections by Using Therapeutic Biomaterials. Polymers 2021, 13, 1556. [Google Scholar] [CrossRef]
- Casillas-Ituarte, N.N.; Staats, A.M.; Lower, B.H.; Stoodley, P.; Lower, S.K. Host blood proteins as bridging ligand in bacterial aggregation as well as anchor point for adhesion in the molecular pathogenesis of Staphylococcus aureus infections. Micron 2021, 150, 103137. [Google Scholar] [CrossRef]
- Krishna, B.V.; Gibb, A.P. Use of octenidine dihydrochloride in meticillin-resistant Staphylococcus aureus decolonisation regimens: A literature review. J. Hosp. Infect. 2010, 74, 199–203. [Google Scholar] [CrossRef]
- Chow, A.; Hon, P.Y.; Tin, G.; Zhang, W.; Poh, B.F.; Ang, B. Intranasal octenidine and universal antiseptic bathing reduce methicillin-resistant Staphylococcus aureus (MRSA) prevalence in extended care facilities. Epidemiol. Infect. 2018, 146, 2036–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F.; Martuscelli, G.; Bellomo, F.; Avtaar Singh, S.S.; Moon, M.R. Infective Endocarditis in High-Income Countries. Metabolites 2022, 12, 682. https://doi.org/10.3390/metabo12080682
Nappi F, Martuscelli G, Bellomo F, Avtaar Singh SS, Moon MR. Infective Endocarditis in High-Income Countries. Metabolites. 2022; 12(8):682. https://doi.org/10.3390/metabo12080682
Chicago/Turabian StyleNappi, Francesco, Giorgia Martuscelli, Francesca Bellomo, Sanjeet Singh Avtaar Singh, and Marc R. Moon. 2022. "Infective Endocarditis in High-Income Countries" Metabolites 12, no. 8: 682. https://doi.org/10.3390/metabo12080682
APA StyleNappi, F., Martuscelli, G., Bellomo, F., Avtaar Singh, S. S., & Moon, M. R. (2022). Infective Endocarditis in High-Income Countries. Metabolites, 12(8), 682. https://doi.org/10.3390/metabo12080682