
O-GlcNAc Modification and Its Role in Diabetic Retinopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
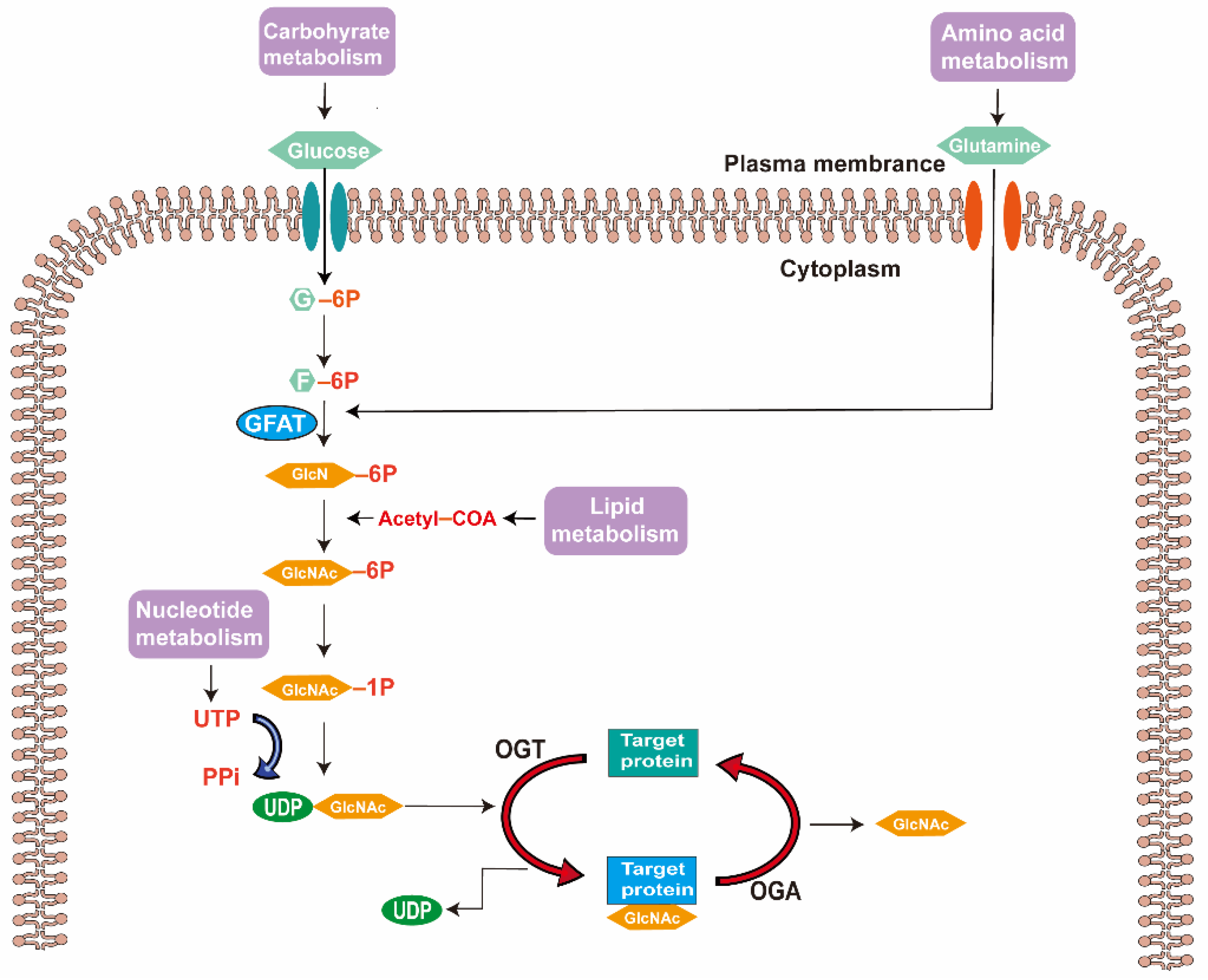
2. O-GlcNAc Modification
2.1. Synthesis of UDP-GlcNAc via Hexosamine Biosynthesis Pathway
2.2. Regulation of O-GlcNAc Modification by OGT and OGA
2.3. Characteristics of O-GlcNAc Modification Compared with Other PTMs
3. Relationship between O-GlcNAc Modification and Diabetes
3.1. Type 2 Diabetes Mellitus and O-GlcNAc Modification
3.2. Type 1 Diabetes Mellitus and O-GlcNAc Modification
4. Relationship between O-GlcNAc Modification and Diabetic Retinopathy
4.1. O-GlcNAc Modification and Retinal Microvascular Lesions
4.1.1. O-GlcNAc Modification and Specialized-Vasculature-Cell Death
4.1.2. O-GlcNAc Modification and Destruction of Endothelial-Cell-Junction Integrity
4.1.3. O-GlcNAc Modification and Neovascularization
4.2. O-GlcNAc Modification and Retinal Neurodegeneration
5. Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Yao, H.Y.; Tseng, K.W.; Nguyen, H.T.; Kuo, C.T.; Wang, H.C. Hyperspectral Ophthalmoscope Images for the Diagnosis of Diabetic Retinopathy Stage. J. Clin. Med. 2020, 9, 1613. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.H.; Fong, K.C. Retinal laser photocoagulation. Med. J. Malays. 2010, 65, 88–94. [Google Scholar]
- Campa, C. New Anti-VEGF Drugs in Ophthalmology. Curr. Drug Targets 2020, 21, 1194–1200. [Google Scholar] [CrossRef]
- Rittiphairoj, T.; Mir, T.A.; Li, T.; Virgili, G. Intravitreal steroids for macular edema in diabetes. Cochrane Database Syst. Rev. 2020, 11, Cd005656. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.H.; Acaba, L.A.; Chenworth, M.L. Surgical Innovations in the Treatment of Diabetic Macular Edema and Diabetic Retinopathy. Curr. Diab Rep. 2019, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Belin, P.J.; Parke, D.W., 3rd. Complications of vitreoretinal surgery. Curr. Opin. Ophthalmol. 2020, 31, 167–173. [Google Scholar] [CrossRef]
- Amin, J.; Sharif, M.; Yasmin, M. A Review on Recent Developments for Detection of Diabetic Retinopathy. Scientifica 2016, 2016, 6838976. [Google Scholar] [CrossRef] [Green Version]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef]
- Gálvez, M.I. Protein kinase C inhibitors in the treatment of diabetic retinopathy. Review. Curr. Pharm. Biotechnol. 2011, 12, 386–391. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role, and therapeutic implications. Redox Biology 2020, 37, 101799. [Google Scholar] [CrossRef] [PubMed]
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galesic, A.; Pratt, M.R. Investigating the Effects of O-GlcNAc Modifications in Parkinson’s Disease Using Semisynthetic α-Synuclein. Methods Mol. Biol. 2020, 2133, 313–326. [Google Scholar] [CrossRef]
- Ferrer, C.M.; Sodi, V.L.; Reginato, M.J. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J. Mol. Biol. 2016, 428, 3282–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhao, X.; Wu, H. Metabolic Stress and Cardiovascular Disease in Diabetes Mellitus: The Role of Protein O-GlcNAc Modification. Arter. Thromb. Vasc. Biol. 2019, 39, 1911–1924. [Google Scholar] [CrossRef]
- Gurel, Z.; Sieg, K.M.; Shallow, K.D.; Sorenson, C.M.; Sheibani, N. Retinal O-linked N-acetylglucosamine protein modifications: Implications for postnatal retinal vascularization and the pathogenesis of diabetic retinopathy. Mol. Vis. 2013, 19, 1047–1059. [Google Scholar]
- Xu, C.; Liu, G.; Liu, X.; Wang, F. O-GlcNAcylation under hypoxic conditions and its effects on the blood-retinal barrier in diabetic retinopathy. Int. J. Mol. Med. 2014, 33, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Wulff-Fuentes, E.; Berendt, R.R.; Massman, L.; Danner, L.; Malard, F.; Vora, J.; Kahsay, R.; Olivier-Van Stichelen, S. The human O-GlcNAcome database and meta-analysis. Sci. Data 2021, 8, 25. [Google Scholar] [CrossRef]
- Zhu, Y.; Hart, G.W. Nutrient regulation of the flow of genetic information by O-GlcNAcylation. Biochem. Soc. Trans. 2021, 49, 867–880. [Google Scholar] [CrossRef]
- Slawson, C.; Lakshmanan, T.; Knapp, S.; Hart, G.W. A mitotic GlcNAcylation/phosphorylation signaling complex alters the posttranslational state of the cytoskeletal protein vimentin. Mol. Biol. Cell 2008, 19, 4130–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butkinaree, C.; Park, K.; Hart, G.W. O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Et Biophys. Acta 2010, 1800, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachara, N.E.; Hart, G.W. O-GlcNAc a sensor of cellular state: The role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta 2004, 1673, 13–28. [Google Scholar] [CrossRef]
- Pravata, V.M.; Muha, V.; Gundogdu, M.; Ferenbach, A.T.; Kakade, P.S.; Vandadi, V.; Wilmes, A.C.; Borodkin, V.S.; Joss, S.; Stavridis, M.P.; et al. Catalytic deficiency of O-GlcNAc transferase leads to X-linked intellectual disability. Proc. Natl. Acad. Sci. USA 2019, 116, 14961–14970. [Google Scholar] [CrossRef] [Green Version]
- Lau, K.S.; Khan, S.; Dennis, J.W. Genome-scale identification of UDP-GlcNAc-dependent pathways. Proteomics 2008, 8, 3294–3302. [Google Scholar] [CrossRef]
- Vasconcelos-Dos-Santos, A.; Oliveira, I.A.; Lucena, M.C.; Mantuano, N.R.; Whelan, S.A.; Dias, W.B.; Todeschini, A.R. Biosynthetic Machinery Involved in Aberrant Glycosylation: Promising Targets for Developing of Drugs Against Cancer. Front. Oncol. 2015, 5, 138. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [CrossRef]
- Hardivillé, S.; Hart, G.W. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.H.; Weng, C.L.; Lin, K.I. O-GlcNAcylation and its role in the immune system. J. Biomed. Sci. 2020, 27, 57. [Google Scholar] [CrossRef]
- Niimi, M.; Ogawara, T.; Yamashita, T.; Yamamoto, Y.; Ueyama, A.; Kambe, T.; Okamoto, T.; Ban, T.; Tamanoi, H.; Ozaki, K.; et al. Identification of GFAT1-L, a novel splice variant of human glutamine: Fructose-6-phosphate amidotransferase (GFAT1) that is expressed abundantly in skeletal muscle. J. Hum. Genet. 2001, 46, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Riesland, L.; Paterson, A.J.; Kudlow, J.E. Phosphorylation of mouse glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2) by cAMP-dependent protein kinase increases the enzyme activity. J. Biol. Chem. 2004, 279, 29988–29993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.L.; Hart, G.W. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J. Biol. Chem. 1994, 269, 19321–19330. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef] [Green Version]
- Shafi, R.; Iyer, S.P.; Ellies, L.G.; O’Donnell, N.; Marek, K.W.; Chui, D.; Hart, G.W.; Marth, J.D. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. USA 2000, 97, 5735–5739. [Google Scholar] [CrossRef] [Green Version]
- Kreppel, L.K.; Blomberg, M.A.; Hart, G.W. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem. 1997, 272, 9308–9315. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef]
- Love, D.C.; Kochan, J.; Cathey, R.L.; Shin, S.H.; Hanover, J.A. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J. Cell Sci. 2003, 116, 647–654. [Google Scholar] [CrossRef] [Green Version]
- Lubas, W.A.; Hanover, J.A. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J. Biol. Chem. 2000, 275, 10983–10988. [Google Scholar] [CrossRef] [Green Version]
- Dierschke, S.K.; Miller, W.P.; Favate, J.S.; Shah, P.; Imamura Kawasawa, Y.; Salzberg, A.C.; Kimball, S.R.; Jefferson, L.S.; Dennis, M.D. O-GlcNAcylation alters the selection of mRNAs for translation and promotes 4E-BP1-dependent mitochondrial dysfunction in the retina. J. Biol. Chem. 2019, 294, 5508–5520. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.D.; Xu, C.; Feng, L.; Wang, F. The augmentation of O-GlcNAcylation reduces glyoxal-induced cell injury by attenuating oxidative stress in human retinal microvascular endothelial cells. Int. J. Mol. Med. 2015, 36, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keembiyehetty, C.N.; Krzeslak, A.; Love, D.C.; Hanover, J.A. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J. Cell Sci. 2011, 124, 2851–2860. [Google Scholar] [CrossRef] [Green Version]
- Wells, L.; Gao, Y.; Mahoney, J.A.; Vosseller, K.; Chen, C.; Rosen, A.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Further characterization of the nucleocytoplasmic beta-N-acetylglucosaminidase, O-GlcNAcase. J. Biol. Chem. 2002, 277, 1755–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, G.W.; Greis, K.D.; Dong, L.Y.; Blomberg, M.A.; Chou, T.Y.; Jiang, M.S.; Roquemore, E.P.; Snow, D.M.; Kreppel, L.K.; Cole, R.N.; et al. O-linked N-acetylglucosamine: The “yin-yang” of Ser/Thr phosphorylation? Nuclear and cytoplasmic glycosylation. Adv. Exp. Med. Biol. 1995, 376, 115–123. [Google Scholar] [PubMed]
- Wang, Z.; Gucek, M.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. USA 2008, 105, 13793–13798. [Google Scholar] [CrossRef] [Green Version]
- Maggio, C.A.; Pi-Sunyer, F.X. Obesity and type 2 diabetes. Endocrinol. Metab. Clin. N. Am. 2003, 32, 805–822. [Google Scholar] [CrossRef]
- George, A.M.; Jacob, A.G.; Fogelfeld, L. Lean diabetes mellitus: An emerging entity in the era of obesity. World J. Diabetes 2015, 6, 613–620. [Google Scholar] [CrossRef]
- Lubas, W.A.; Frank, D.W.; Krause, M.; Hanover, J.A. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J. Biol. Chem. 1997, 272, 9316–9324. [Google Scholar] [CrossRef] [Green Version]
- Baumann, D.; Wong, A.; Akhaphong, B.; Jo, S.; Pritchard, S.; Mohan, R.; Chung, G.; Zhang, Y.; Alejandro, E.U. Role of nutrient-driven O-GlcNAc-post-translational modification in pancreatic exocrine and endocrine islet development. Development 2020, 147, dev186643. [Google Scholar] [CrossRef]
- Ida, S.; Morino, K.; Sekine, O.; Ohashi, N.; Kume, S.; Chano, T.; Iwasaki, K.; Harada, N.; Inagaki, N.; Ugi, S.; et al. Diverse metabolic effects of O-GlcNAcylation in the pancreas but limited effects in insulin-sensitive organs in mice. Diabetologia 2017, 60, 1761–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, S.A.; Lane, M.D.; Hart, G.W. Regulation of the O-linked beta-N-acetylglucosamine transferase by insulin signaling. J. Biol. Chem. 2008, 283, 21411–21417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Häring, H.U. The impact of insulin resistance on the kidney and vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Garabadu, D.; Krishnamurthy, S. Metformin attenuates hepatic insulin resistance in type-2 diabetic rats through PI3K/Akt/GLUT-4 signalling independent to bicuculline-sensitive GABA(A) receptor stimulation. Pharm. Biol. 2017, 55, 722–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buse, M.G. Hexosamines, insulin resistance, and the complications of diabetes: Current status. Am. J. Physiology. Endocrinol. Metab. 2006, 290, E1–E8. [Google Scholar] [CrossRef]
- Sesti, G.; Federici, M.; Hribal, M.L.; Lauro, D.; Sbraccia, P.; Lauro, R. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. Faseb J. 2001, 15, 2099–2111. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Ryu, J.; Lee, W. O-GlcNAc modification on IRS-1 and Akt2 by PUGNAc inhibits their phosphorylation and induces insulin resistance in rat primary adipocytes. Exp. Mol. Med. 2005, 37, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Kaleem, A.; Javed, S.; Rehman, N.; Abdullah, R.; Iqtedar, M.; Aftab, M.N.; Hoessli, D.C.; Haq, I.U. Phosphorylated and O-GlcNAc Modified IRS-1 (Ser1101) and -2 (Ser1149) Contribute to Human Diabetes Type II. Protein Pept. Lett. 2021, 28, 333–339. [Google Scholar] [CrossRef]
- Whelan, S.A.; Dias, W.B.; Thiruneelakantapillai, L.; Lane, M.D.; Hart, G.W. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-Linked beta-N-acetylglucosamine in 3T3-L1 adipocytes. J. Biol. Chem. 2010, 285, 5204–5211. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ongusaha, P.P.; Miles, P.D.; Havstad, J.C.; Zhang, F.; So, W.V.; Kudlow, J.E.; Michell, R.H.; Olefsky, J.M.; Field, S.J.; et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008, 451, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Miyazaki, J.; Hart, G.W. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch. Biochem. Biophys. 2003, 415, 155–163. [Google Scholar] [CrossRef]
- Kebede, M.; Ferdaoussi, M.; Mancini, A.; Alquier, T.; Kulkarni, R.N.; Walker, M.D.; Poitout, V. Glucose activates free fatty acid receptor 1 gene transcription via phosphatidylinositol-3-kinase-dependent O-GlcNAcylation of pancreas-duodenum homeobox-1. Proc. Natl. Acad. Sci. USA 2012, 109, 2376–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasumi, K.; Esaki, R.; Iwachidow, K.; Yasuhara, Y.; Ogi, K.; Tanaka, H.; Nakata, M.; Yano, T.; Shimakawa, K.; Taketomi, S.; et al. Overexpression of GPR40 in pancreatic beta-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes 2009, 58, 1067–1076. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [Green Version]
- Filhoulaud, G.; Benhamed, F.; Pagesy, P.; Bonner, C.; Fardini, Y.; Ilias, A.; Movassat, J.; Burnol, A.F.; Guilmeau, S.; Kerr-Conte, J.; et al. O-GlcNacylation Links TxNIP to Inflammasome Activation in Pancreatic β Cells. Front. Endocrinol. 2019, 10, 291. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.S.; Han, D.; Park, J.; Kwak, T.K.; Oh, M.A.; Lee, S.A.; Choi, S.; Park, Z.Y.; Kim, Y.; Lee, J.W. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Exp. Cell Res. 2008, 314, 2238–2248. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandris, C.; Andreozzi, F.; Federici, M.; Cardellini, M.; Brunetti, A.; Ranalli, M.; Del Guerra, S.; Lauro, D.; Del Prato, S.; Marchetti, P.; et al. Increased O-glycosylation of insulin signaling proteins results in their impaired activation and enhanced susceptibility to apoptosis in pancreatic beta-cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 959–961. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.; Faria de Abreu, J.R.; Campos, A.J. Early breakdown of the blood-retinal barrier in diabetes. Br. J. Ophthalmol. 1975, 59, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, D.A.; Keane, P.A.; Rajendram, R.; Karampelas, M.; Selvam, S.; Powner, M.B.; Fruttiger, M.; Tufail, A.; Egan, C.A. Patterns of peripheral retinal and central macula ischemia in diabetic retinopathy as evaluated by ultra-widefield fluorescein angiography. Am. J. Ophthalmol. 2014, 158, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.K.; Abràmoff, M.D. Diabetic retinopathy is a neurodegenerative disorder. Vis. Res. 2017, 139, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, K.B.; Frydkjaer-Olsen, U.; Grauslund, J. Vascular Changes and Neurodegeneration in the Early Stages of Diabetic Retinopathy: Which Comes First? Ophthalmic Res. 2016, 56, 1–9. [Google Scholar] [CrossRef]
- Gaceb, A.; Paul, G. Pericyte Secretome. Adv. Exp. Med. Biol. 2018, 1109, 139–163. [Google Scholar] [CrossRef]
- Frank, R.N.; Dutta, S.; Mancini, M.A. Pericyte coverage is greater in the retinal than in the cerebral capillaries of the rat. Investig. Ophthalmol. Vis. Sci. 1987, 28, 1086–1091. [Google Scholar]
- Gurel, Z.; Sheibani, N. O-Linked β-N-acetylglucosamine (O-GlcNAc) modification: A new pathway to decode pathogenesis of diabetic retinopathy. Clin. Sci. 2018, 132, 185–198. [Google Scholar] [CrossRef]
- Gurel, Z.; Zaro, B.W.; Pratt, M.R.; Sheibani, N. Identification of O-GlcNAc modification targets in mouse retinal pericytes: Implication of p53 in pathogenesis of diabetic retinopathy. PLoS ONE 2014, 9, e95561. [Google Scholar] [CrossRef]
- Yang, W.H.; Kim, J.E.; Nam, H.W.; Ju, J.W.; Kim, H.S.; Kim, Y.S.; Cho, J.W. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat. Cell Biol. 2006, 8, 1074–1083. [Google Scholar] [CrossRef]
- Ghimire, K.; Altmann, H.M.; Straub, A.C.; Isenberg, J.S. Nitric oxide: What’s new to NO? Am. J. Physiol. Cell Physiol. 2017, 312, C254–C262. [Google Scholar] [CrossRef]
- Aulak, K.S.; Barnes, J.W.; Tian, L.; Mellor, N.E.; Haque, M.M.; Willard, B.; Li, L.; Comhair, S.C.; Stuehr, D.J.; Dweik, R.A. Specific O-GlcNAc modification at Ser-615 modulates eNOS function. Redox Biol. 2020, 36, 101625. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Taguchi, T.; Matsumura, T.; Pestell, R.; Edelstein, D.; Giardino, I.; Suske, G.; Rabbani, N.; Thornalley, P.J.; Sarthy, V.P.; et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J. Biol. Chem. 2007, 282, 31038–31045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, A.; Eshwaran, R.; Poschet, G.; Lomada, S.; Halawa, M.; Wilhelm, K.; Schmidt, M.; Hammes, H.P.; Wieland, T.; Feng, Y. Involvement of NDPK-B in Glucose Metabolism-Mediated Endothelial Damage via Activation of the Hexosamine Biosynthesis Pathway and Suppression of O-GlcNAcase Activity. Cells 2020, 9, 2324. [Google Scholar] [CrossRef] [PubMed]
- Gardizi, M.; Kurschat, C.; Riese, A.; Hahn, M.; Krieg, T.; Mauch, C.; Kurschat, P. A decreased ratio between serum levels of the antagonistic angiopoietins 1 and 2 indicates tumour progression of malignant melanoma. Arch. Dermatol. Res. 2012, 304, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donzé, O.; Lin, T.A.; Lawrence, J.C., Jr.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Z.; Li, L.; Gong, W.; Lazenby, A.J.; Swanson, B.J.; Herring, L.E.; Asara, J.M.; Singer, J.D.; Wen, H. Myeloid-derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J. Exp. Med. 2017, 214, 1093–1109. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Liu, G.D.; Feng, L.; Zhang, C.H.; Wang, F. Identification of O-GlcNAcylation Modification in Diabetic Retinopathy and Crosstalk with Phosphorylation of STAT3 in Retina Vascular Endothelium Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 1389–1402. [Google Scholar] [CrossRef]
- Lenin, R.; Nagy, P.G.; Jha, K.A.; Gangaraju, R. GRP78 translocation to the cell surface and O-GlcNAcylation of VE-Cadherin contribute to ER stress-mediated endothelial permeability. Sci. Rep. 2019, 9, 10783. [Google Scholar] [CrossRef]
- Le, Y.Z. VEGF production and signaling in Müller glia are critical to modulating vascular function and neuronal integrity in diabetic retinopathy and hypoxic retinal vascular diseases. Vision Res. 2017, 139, 108–114. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065–15075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, K.; Alekseev, O.; Qi, X.; Cho, W.; Azizkhan-Clifford, J. O-GlcNAc modification of transcription factor Sp1 mediates hyperglycemia-induced VEGF-A upregulation in retinal cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7862–7873. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.D.; Oh, D.J.; Wong, L.L.; Amarnani, D.; Park-Windhol, C.; Sanchez, A.V.; Cardona-Velez, J.; McGuone, D.; Stemmer-Rachamimov, A.O.; Eliott, D.; et al. Identification of RUNX1 as a Mediator of Aberrant Retinal Angiogenesis. Diabetes 2017, 66, 1950–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, X.; Wang, H.; Niu, T.; Jiang, Y.; Shi, X.; Liu, K. RUNX1 can mediate the glucose and O-GlcNAc-driven proliferation and migration of human retinal microvascular endothelial cells. BMJ Open Diabetes Res. Care 2021, 9, e001898. [Google Scholar] [CrossRef] [PubMed]
- Haefliger, J.A.; Allagnat, F.; Hamard, L.; Le Gal, L.; Meda, P.; Nardelli-Haefliger, D.; Génot, E.; Alonso, F. Targeting Cx40 (Connexin40) Expression or Function Reduces Angiogenesis in the Developing Mouse Retina. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2136–2146. [Google Scholar] [CrossRef] [Green Version]
- Makino, A.; Dai, A.; Han, Y.; Youssef, K.D.; Wang, W.; Donthamsetty, R.; Scott, B.T.; Wang, H.; Dillmann, W.H. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am. J. Physiol. Cell Physiol. 2015, 309, C593–C599. [Google Scholar] [CrossRef] [Green Version]
- Makino, A.; Platoshyn, O.; Suarez, J.; Yuan, J.X.; Dillmann, W.H. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am. J. Physiol. Cell Physiol. 2008, 295, C221–C230. [Google Scholar] [CrossRef] [Green Version]
- Zafar, S.; Sachdeva, M.; Frankfort, B.J.; Channa, R. Retinal Neurodegeneration as an Early Manifestation of Diabetic Eye Disease and Potential Neuroprotective Therapies. Curr. Diabetes Rep. 2019, 19, 17. [Google Scholar] [CrossRef]
- van Dijk, H.W.; Verbraak, F.D.; Stehouwer, M.; Kok, P.H.; Garvin, M.K.; Sonka, M.; DeVries, J.H.; Schlingemann, R.O.; Abràmoff, M.D. Association of visual function and ganglion cell layer thickness in patients with diabetes mellitus type 1 and no or minimal diabetic retinopathy. Vision Res. 2011, 51, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Park, J.W.; Park, S.J.; Kim, K.Y.; Chung, J.W.; Chun, M.H.; Oh, S.J. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia 2003, 46, 1260–1268. [Google Scholar] [CrossRef]
- Dai, W.; Dierschke, S.K.; Toro, A.L.; Dennis, M.D. Consumption of a high fat diet promotes protein O-GlcNAcylation in mouse retina via NR4A1-dependent GFAT2 expression. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3568–3576. [Google Scholar] [CrossRef] [PubMed]
- Križaj, D. Polymodal Sensory Integration in Retinal Ganglion Cells. Adv. Exp. Med. Biol. 2016, 854, 693–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, T.S.; Barber, A.J. Retinal ganglion cells in diabetes. J. Physiol. 2008, 586, 4401–4408. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, M.J.; Choi, M.Y.; Kim, Y.S.; Yoo, J.M.; Hong, E.K.; Ju, S.; Choi, W.S. Aralia elata inhibits neurodegeneration by downregulating O-GlcNAcylation of NF-κB in diabetic mice. Int. J. Ophthalmol. 2017, 10, 1203–1211. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, M.; Choi, M.Y.; Lee, D.H.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Kim, S.J.; Yoo, J.M.; et al. Metformin protects against retinal cell death in diabetic mice. Biochem. Biophys. Res. Commun. 2017, 492, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, M.; Zhao, S.; Lu, Q.; Ni, L.; Zou, C.; Lu, L.; Xu, X.; Guan, H.; Zheng, Z.; et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: A novel inhibitory effect of minocycline. Inflamm. Res. Off. J. Eur. Histamine Res. Society 2017, 66, 157–166. [Google Scholar] [CrossRef]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef]
- Zhou, J.; Shen, X.; Lu, Q.; Zhang, M. Thioredoxin-Interacting Protein (TXNIP) Suppresses Expression of Glutamine Synthetase by Inducing Oxidative Stress in Retinal Muller Glia Under Diabetic Conditions. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 1460–1466. [Google Scholar] [CrossRef] [Green Version]
- Singh, L.P. Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J. Clin. Exp. Ophthalmol. 2013, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Ao, H.; Li, H.; Zhao, X.; Liu, B.; Lu, L. TXNIP positively regulates the autophagy and apoptosis in the rat müller cell of diabetic retinopathy. Life Sci. 2021, 267, 118988. [Google Scholar] [CrossRef]
- Devi, T.S.; Hosoya, K.; Terasaki, T.; Singh, L.P. Critical role of TXNIP in oxidative stress, DNA damage and retinal pericyte apoptosis under high glucose: Implications for diabetic retinopathy. Exp. Cell Res. 2013, 319, 1001–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-specific GlcNAcylation of human erythrocyte proteins: Potential biomarker(s) for diabetes. Diabetes 2009, 58, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myslicki, J.P.; Shearer, J.; Hittel, D.S.; Hughey, C.C.; Belke, D.D. O-GlcNAc modification is associated with insulin sensitivity in the whole blood of healthy young adult males. Diabetol. Metab. Syndr. 2014, 6, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierschke, S.K.; Toro, A.L.; Barber, A.J.; Arnold, A.C.; Dennis, M.D. Angiotensin-(1-7) Attenuates Protein O-GlcNAcylation in the Retina by EPAC/Rap1-Dependent Inhibition of O-GlcNAc Transferase. Investig. Ophthalmol. Vis. Sci. 2020, 61, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Shan, Z.; Lei, B.; Yuan, L.; Liu, X.; Nakagawa, T.; Grant, M.B.; Lewin, A.S.; Hauswirth, W.W.; Raizada, M.K.; et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Abramowitz, L.K.; Harly, C.; Das, A.; Bhandoola, A.; Hanover, J.A. Blocked O-GlcNAc cycling disrupts mouse hematopoeitic stem cell maintenance and early T cell development. Sci. Rep. 2019, 9, 12569. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Dong, W.; Li, J.; Kong, Y.; Ren, X. O-GlcNAc Modification and Its Role in Diabetic Retinopathy. Metabolites 2022, 12, 725. https://doi.org/10.3390/metabo12080725
Liu C, Dong W, Li J, Kong Y, Ren X. O-GlcNAc Modification and Its Role in Diabetic Retinopathy. Metabolites. 2022; 12(8):725. https://doi.org/10.3390/metabo12080725
Chicago/Turabian StyleLiu, Chengzhi, Wenkang Dong, Jun Li, Ying Kong, and Xiang Ren. 2022. "O-GlcNAc Modification and Its Role in Diabetic Retinopathy" Metabolites 12, no. 8: 725. https://doi.org/10.3390/metabo12080725
APA StyleLiu, C., Dong, W., Li, J., Kong, Y., & Ren, X. (2022). O-GlcNAc Modification and Its Role in Diabetic Retinopathy. Metabolites, 12(8), 725. https://doi.org/10.3390/metabo12080725