
Marine-Derived Compounds for CDK5 Inhibition in Cancer: Integrating Multi-Stage Virtual Screening, MM/GBSA Analysis and Molecular Dynamics Investigations

 , , , , , ,
, , , , , ,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Ligand Preparation
2.3. Molecular Docking
2.4. MM/GBSA Binding Free Energy Calculation
2.5. ADME and Toxicity Prediction
2.6. Molecular Dynamics Simulation
3. Results and Discussion
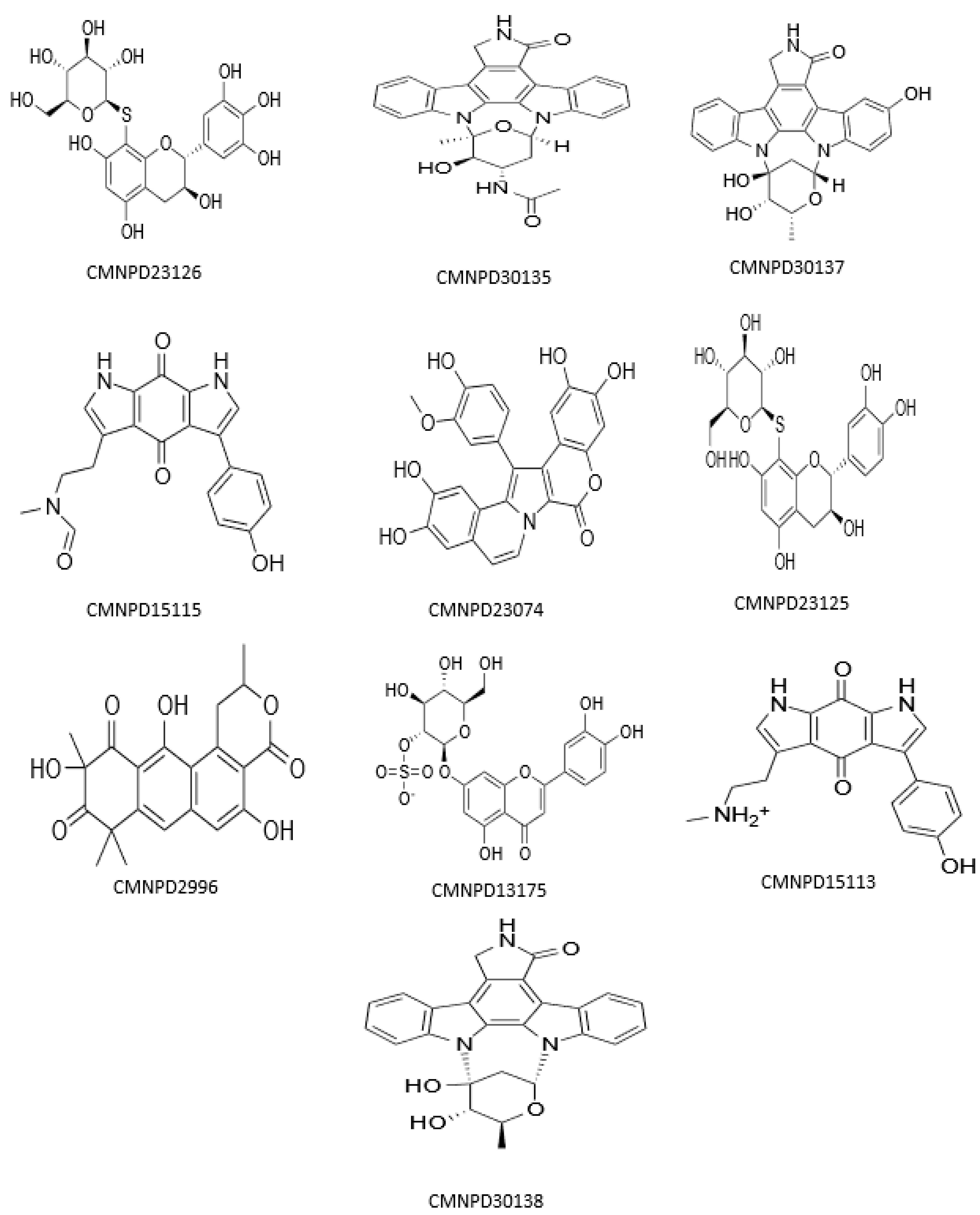
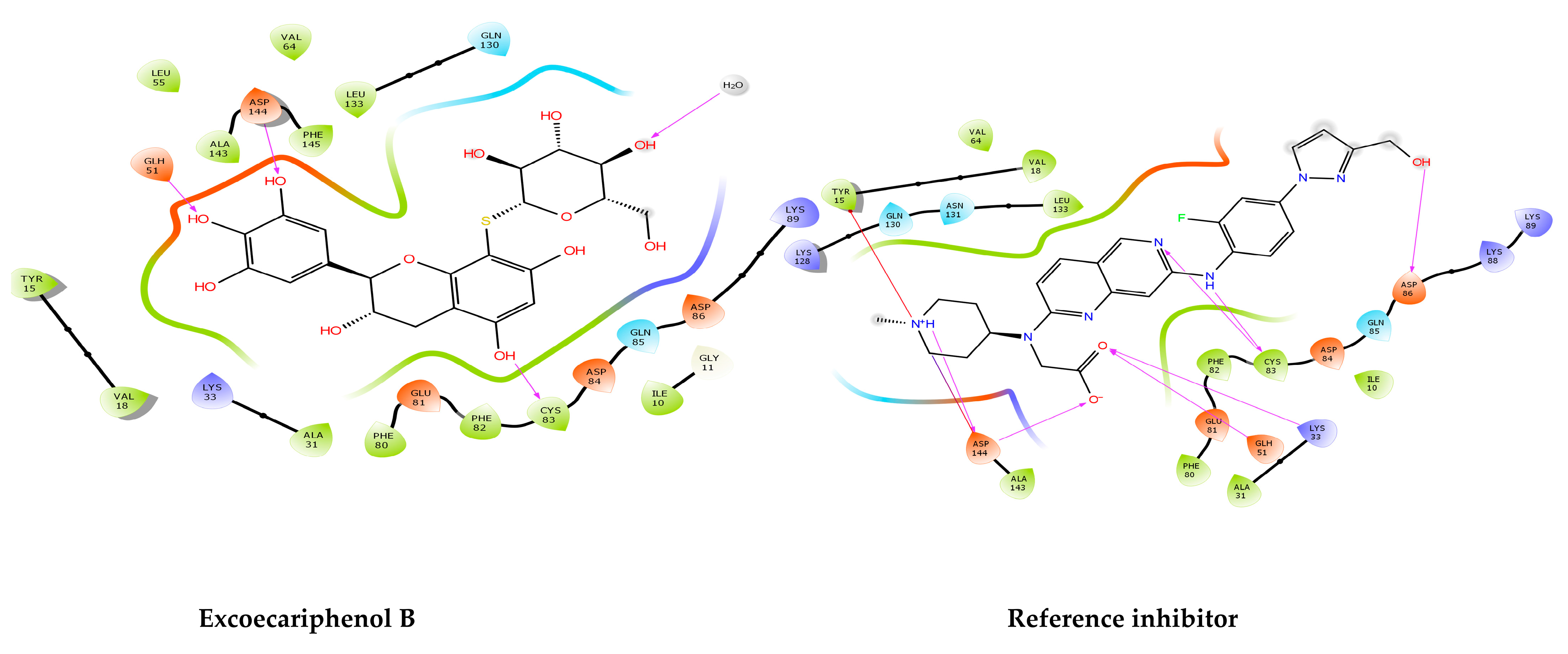
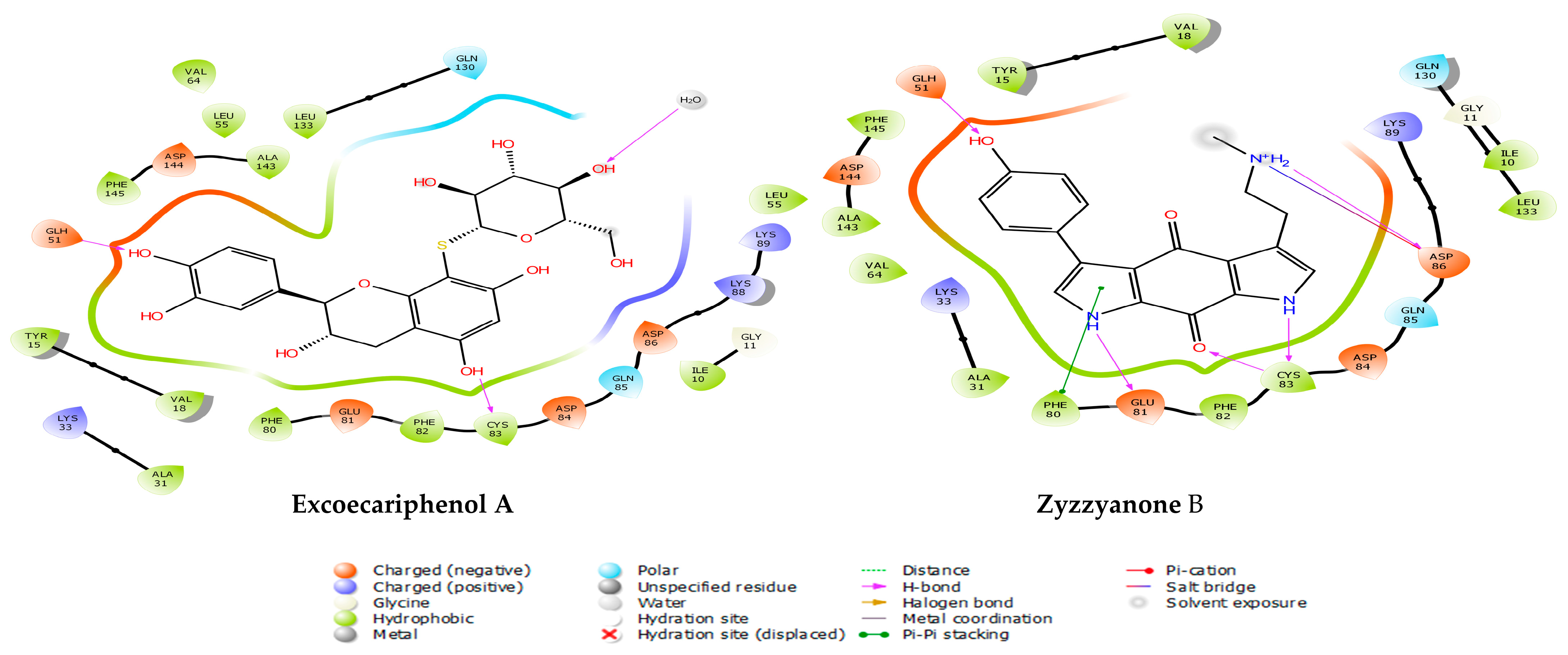
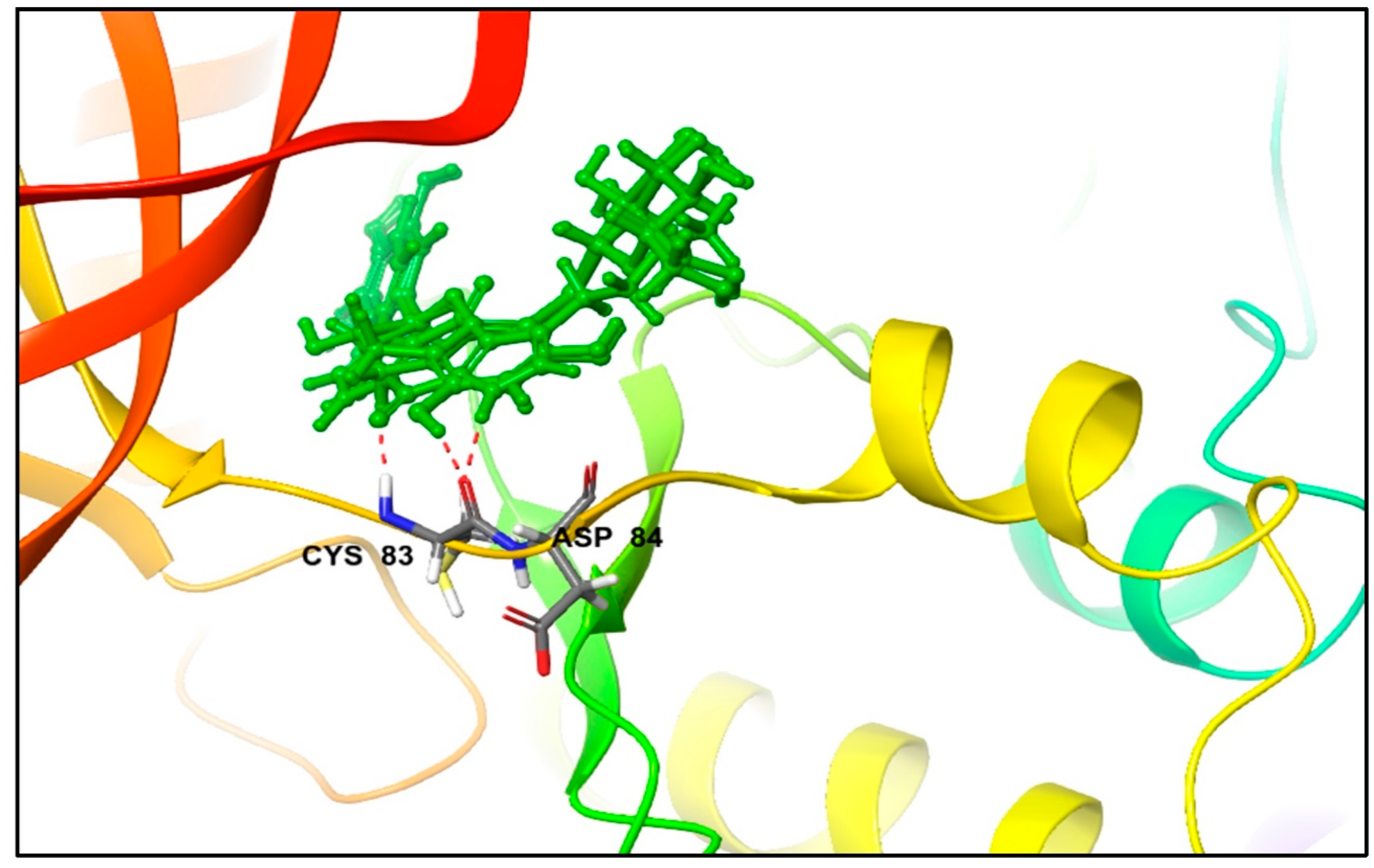
3.1. Molecular Docking and MM/GBSA Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | Name | Docking Score | XP G Score | MM/GBSA dG Bind | Taxonomy |
|---|---|---|---|---|---|
| CMNPD23126 | excoecariphenol B | −13.343 | −13.343 | −83.78 | Excoecaria agallocha [54] |
| Co-crystallized reference | 2-[[7-[[2-fluoranyl-4-[3-(hydroxymethyl) pyrazol-1-yl] phenyl] amino]-1,6-naphthyridin-2-yl]-(1-methylpiperidin-4-yl) amino] ethanoic acid | −13.516 | −13.516 | −74.93 | - |
| CMNPD23125 | excoecariphenol A | −12.018 | −12.018 | −71.58 | Excoecaria agallocha [54] |
| CMNPD15113 | zyzzyanone B | −12.791 | −12.791 | −71.05 | Zyzzya fuliginosa [55,56] |
| CMNPD15115 | zyzzyanone D | −12.058 | −12.058 | −69.36 | Zyzzya fuliginosa [55,56] |
| CMNPD23074 | lamellarin A5 | −12.015 | −12.015 | −61.95 | Didemnum species [57] |
| CMNPD30137 | streptocarbazole E | −13.863 | −13.863 | −60.50 | Streptomyces species [58] |
| CMNPD30135 | 3′-O-demethyl-4′-N-demethyl-4′-N-acetyl-4′-epi-staurosporine | −12.547 | −12.547 | −56.15 | Streptomyces species [59] |
| CMNPD30138 | streptocarbazole C | −12.190 | −12.190 | −53.82 | Streptomyces species [58] |
| CMNPD13175 | thalassiolin A | −12.310 | −12.310 | −51.22 | Thalassia testudinum [60] |
| CMNPD2996 | 2-hydroxygarvin B | −12.449 | −12.449 | −47.20 | Garveia annulata [61] |
3.2. ADME and Toxicity Analysis
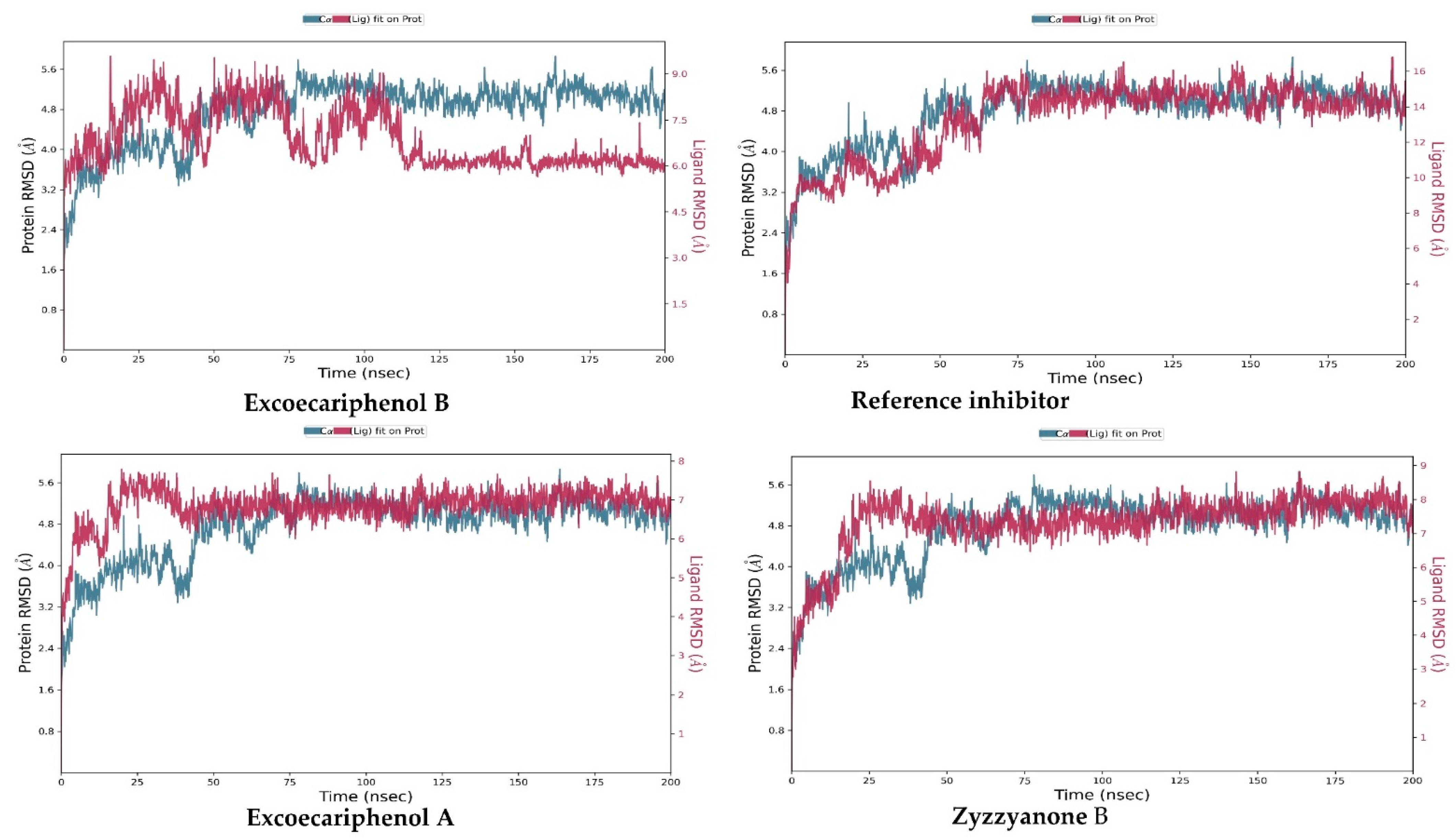
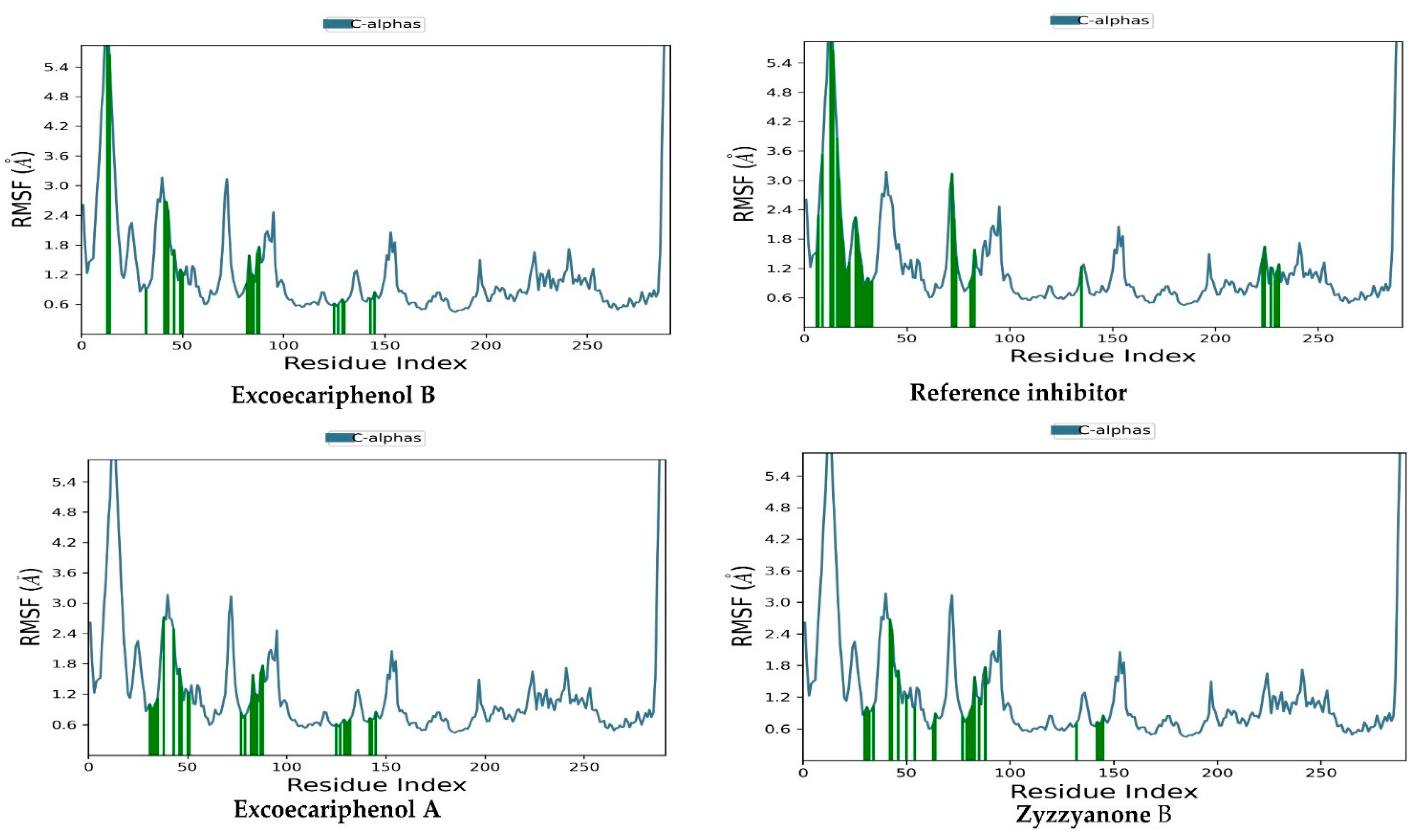
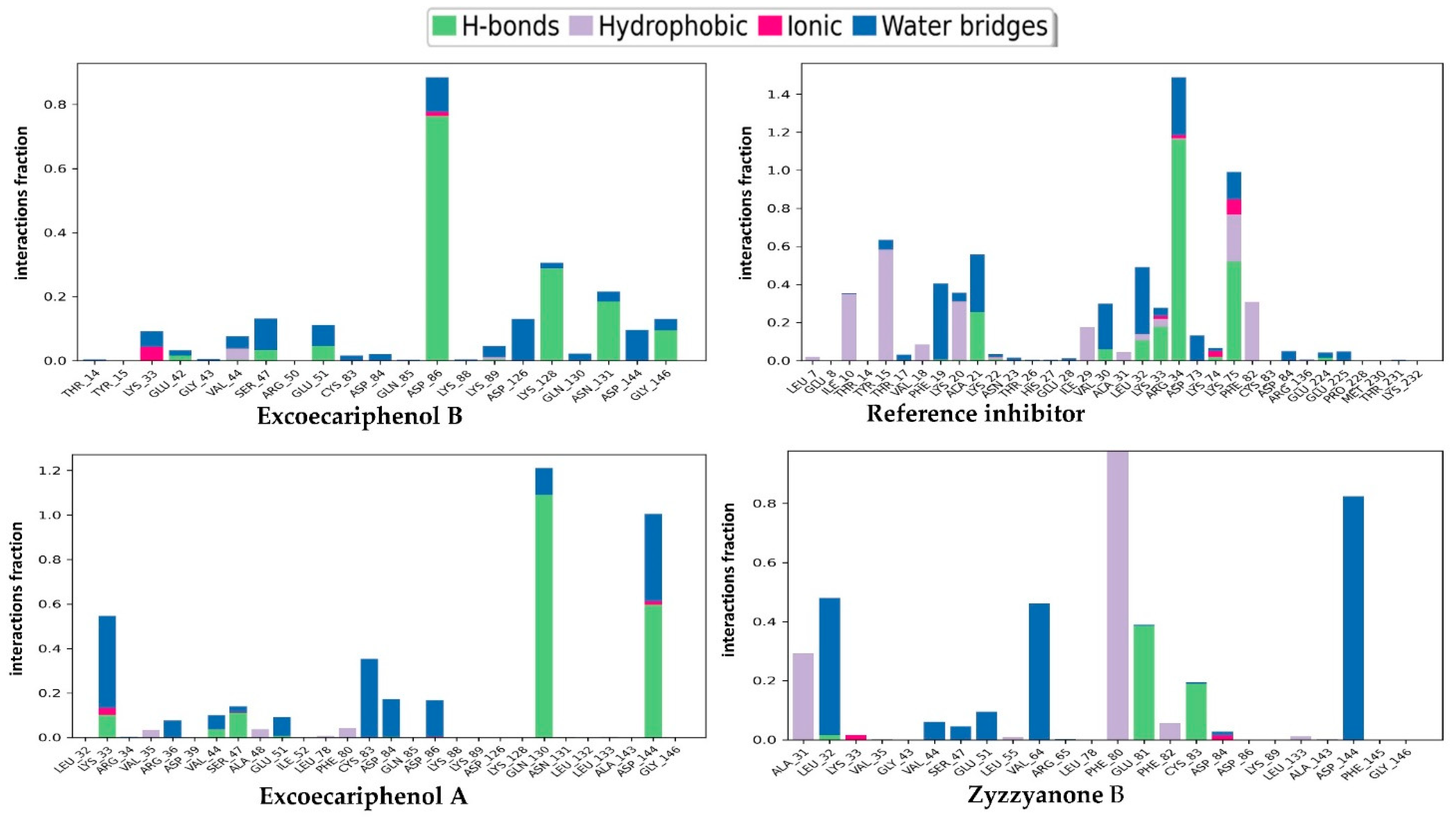
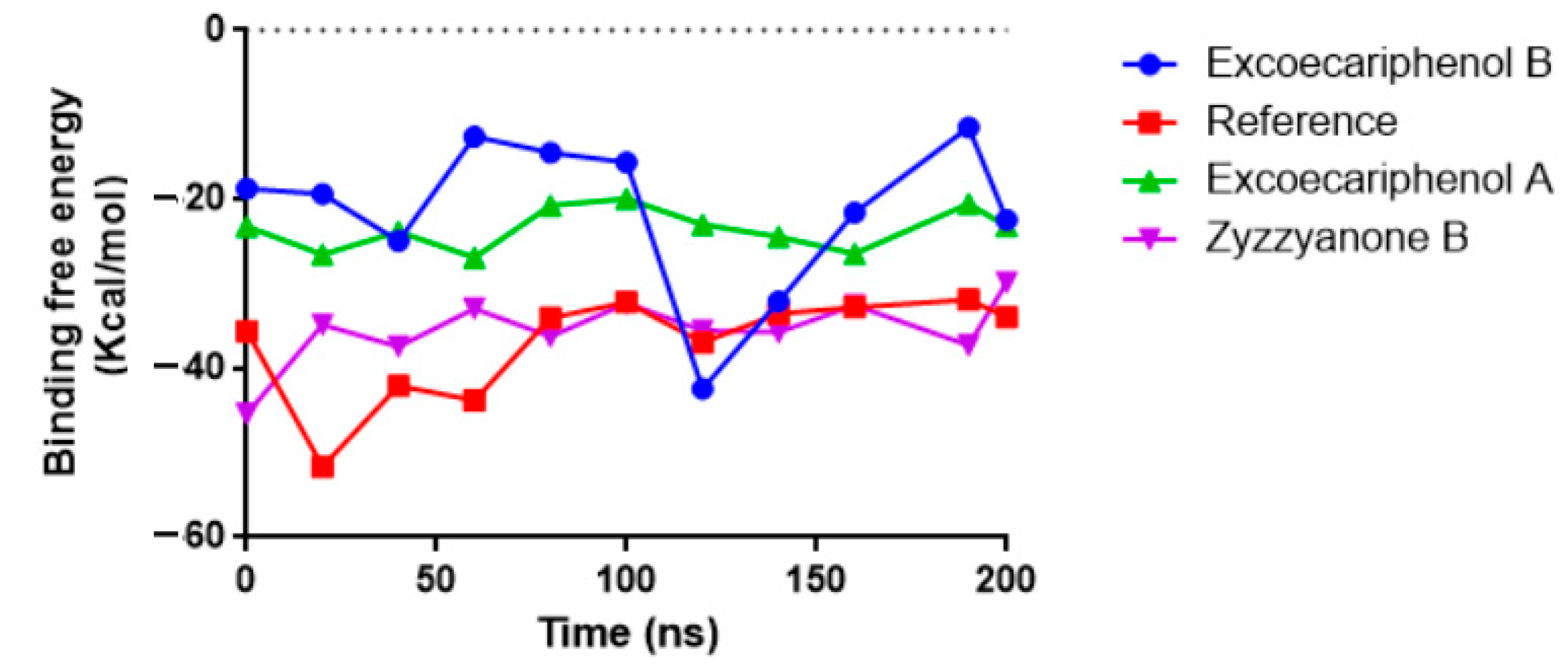
3.3. Molecular Dynamics (MD) Simulations Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| CDK5 | Cyclin-dependent kinase 5 |
| CHD | Coronary heart disease |
| CMNPD | Comprehensive marine natural product database |
| FDA | Food and Drug Administration |
| HTVS | High-throughput virtual screening |
| MD | Molecular dynamics |
| MM/GBSA | Molecular mechanics/generalized born surface area |
| NPT | Isothermal–isobaric |
| OPLS | Optimized potentials for liquid simulations |
| RMSD | Root means square deviation |
| RMSF | Root mean square fluctuation |
| TIP3P | Transferable interaction potential |
| PDB | Protein data bank |
| SP | Standard precision |
| XP | Extra precision |
References
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tan, V.B.C.; Lim, K.M.; Tay, T.E. The Activation and Inhibition of Cyclin-Dependent Kinase-5 by Phosphorylation. Biochemistry 2007, 46, 10841–10851. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, M.R.; Pant, H.C.; Wada, E.; Battey, J.F. Neuronal Cdc2-like Kinase: A Cdc2-Related Protein Kinase with Predominantly Neuronal Expression. Proc. Natl. Acad. Sci. USA 1992, 89, 10867–10871. [Google Scholar] [CrossRef] [PubMed]
- Enserink, J.M.; Chymkowitch, P. Cell Cycle-Dependent Transcription: The Cyclin Dependent Kinase Cdk1 Is a Direct Regulator of Basal Transcription Machineries. Int. J. Mol. Sci. 2022, 23, 1293. [Google Scholar] [CrossRef]
- Futatsugi, A.; Utreras, E.; Rudrabhatla, P.; Jaffe, H.; Pant, H.C.; Kulkarni, A.B. Cyclin-Dependent Kinase 5 Regulates E2F Transcription Factor through Phosphorylation of Rb Protein in Neurons. Cell Cycle 2012, 11, 1603–1610. [Google Scholar] [CrossRef]
- Gonzalez, L.; Domingo-Muelas, A.; Duart-Abadia, P.; Nuñez, M.; Mikolcevic, P.; Llonch, E.; Cubillos-Rojas, M.; Cánovas, B.; Forrow, S.M.A.; Morante-Redolat, J.M.; et al. The Atypical CDK Activator RingoA/Spy1 Regulates Exit from Quiescence in Neural Stem Cells. iScience 2023, 26, 106202. [Google Scholar] [CrossRef]
- Otyepka, M.; Bártová, I.; Kříž, Z.; Koča, J. Different Mechanisms of CDK5 and CDK2 Activation as Revealed by CDK5/P25 and CDK2/Cyclin A Dynamics. J. Biol. Chem. 2006, 281, 7271–7281. [Google Scholar] [CrossRef]
- Tarricone, C.; Dhavan, R.; Peng, J.; Areces, L.B.; Tsai, L.-H.; Musacchio, A. Structure and Regulation of the CDK5-P25nck5a Complex. Mol. Cell 2001, 8, 657–669. [Google Scholar] [CrossRef]
- Semenov, I.; Akyuz, C.; Roginskaya, V.; Chauhan, D.; Corey, S.J. Growth Inhibition and Apoptosis of Myeloma Cells by the CDK Inhibitor Flavopiridol. Leuk. Res. 2002, 26, 271–280. [Google Scholar] [CrossRef]
- Bei, Y.; Cheng, N.; Chen, T.; Shu, Y.; Yang, Y.; Yang, N.; Zhou, X.; Liu, B.; Wei, J.; Liu, Q.; et al. CDK5 Inhibition Abrogates TNBC Stem-Cell Property and Enhances Anti-PD-1 Therapy. Adv. Sci. 2020, 7, 2001417. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Holvey-Bates, E.G.; Mahen, K.; Willard, B.; Stark, G.R. The Ubiquitin E3 Ligase FBXO22 Degrades PD-L1 and Sensitizes Cancer Cells to DNA Damage. Proc. Natl. Acad. Sci. USA 2021, 118, e2112674118. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tan, Z.; Li, H.; Lin, M.; Jiang, Y.; Liang, L.; Ma, Q.; Gou, J.; Ning, L.; Li, X.; et al. Melatonin Reduces Proliferation and Promotes Apoptosis of Bladder Cancer Cells by Suppressing O-GlcNAcylation of Cyclin-Dependent-like Kinase 5. J. Pineal Res. 2021, 71, e12765. [Google Scholar] [CrossRef]
- Feldmann, G.; Mishra, A.; Hong, S.M.; Bisht, S.; Strock, C.J.; Ball, D.W.; Goggins, M.; Maitra, A.; Nelkin, B.D. Inhibiting the Cyclin-Dependent Kinase CDK5 Blocks Pancreatic Cancer Formation and Progression through the Suppression of Ras-Ral Signaling. Cancer Res. 2010, 70, 4460–4469. [Google Scholar] [CrossRef]
- Huang, J.; Chen, P.; Liu, K.; Liu, J.; Zhou, B.; Wu, R.; Peng, Q.; Liu, Z.X.; Li, C.; Kroemer, G.; et al. CDK1/2/5 Inhibition Overcomes IFNG-Mediated Adaptive Immune Resistance in Pancreatic Cancer. Gut 2021, 70, 890–899. [Google Scholar] [CrossRef]
- Lindqvist, J.; Imanishi, S.Y.; Torvaldson, E.; Malinen, M.; Remes, M.; Örn, F.; Palvimo, J.J.; Eriksson, J.E. Cyclin-Dependent Kinase 5 Acts as a Critical Determinant of AKT-Dependent Proliferation and Regulates Differential Gene Expression by the Androgen Receptor in Prostate Cancer Cells. Mol. Biol. Cell 2015, 26, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, P.; Zhang, C.; Chiewchengchol, D.; Zhao, F.; Yu, H.; Li, J.; Kambara, H.; Luo, K.Y.; Venkataraman, A.; et al. Positive Regulation of Interleukin-1β Bioactivity by Physiological ROS-Mediated Cysteine S-Glutathionylation. Cell Rep. 2017, 20, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Xie, F.F.; Zhu, X.J.; Lin, F.; Pan, S.S.; Gong, L.H.; Qiu, J.G.; Zhang, W.J.; Jiang, Q.W.; Mei, X.L.; et al. Cyclin-Dependent Kinase Inhibitor Dinaciclib Potently Synergizes with Cisplatin in Preclinical Models of Ovarian Cancer. Oncotarget 2015, 6, 14926–14939. [Google Scholar] [CrossRef]
- Ehrlich, S.M.; Liebl, J.; Ardelt, M.A.; Lehr, T.; De Toni, E.N.; Mayr, D.; Brandl, L.; Kirchner, T.; Zahler, S.; Gerbes, A.L.; et al. Targeting Cyclin Dependent Kinase 5 in Hepatocellular Carcinoma—A Novel Therapeutic Approach. J. Hepatol. 2015, 63, 102–113. [Google Scholar] [CrossRef]
- Pozo, K.; Castro-Rivera, E.; Tan, C.; Plattner, F.; Schwach, G.; Siegl, V.; Meyer, D.; Guo, A.; Gundara, J.; Mettlach, G.; et al. The Role of Cdk5 in Neuroendocrine Thyroid Cancer. Cancer Cell 2013, 24, 499–511. [Google Scholar] [CrossRef]
- Sun, S.-S.; Zhou, X.; Huang, Y.-Y.; Kong, L.-P.; Mei, M.; Guo, W.-Y.; Zhao, M.-H.; Ren, Y.; Shen, Q.; Zhang, L. Targeting STAT3/MiR-21 Axis Inhibits Epithelial-Mesenchymal Transition via Regulating CDK5 in Head and Neck Squamous Cell Carcinoma. Mol. Cancer 2015, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Lin, C.; Yu, Q.; Zhang, D.; Liu, Y.; Zhang, L.; Zhou, Z.; Zhang, J.; Ouyang, L. Novel Medicinal Chemistry Strategies Targeting CDK5 for Drug Discovery. J. Med. Chem. 2023, 66, 7140–7161. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.H.; Malojcic, G.; Clugston, S.L.; Williams, B.; Coeffet-Le Gal, M.; Pan-Zhou, X.R.; Venkatachalan, S.; Harmange, J.C.; Ledeboer, M. Discovery and Optimization of Highly Selective Inhibitors of CDK5. J. Med. Chem. 2022, 65, 3575–3596. [Google Scholar] [CrossRef]
- Senderowicz, A.M.; Headlee, D.; Stinson, S.F.; Lush, R.M.; Kalil, N.; Villalba, L.; Hill, K.; Steinberg, S.M.; Figg, W.D.; Tompkins, A.; et al. Phase I Trial of Continuous Infusion Flavopiridol, a Novel Cyclin-Dependent Kinase Inhibitor, in Patients with Refractory Neoplasms. J. Clin. Oncol. 1998, 16, 2986–2999. [Google Scholar] [CrossRef]
- Tan, A.R.; Yang, X.; Berman, A.; Zhai, S.; Sparreboom, A.; Parr, A.L.; Chow, C.; Brahim, J.S.; Steinberg, S.M.; Figg, W.D.; et al. Phase I Trial of the Cyclin-Dependent Kinase Inhibitor Flavopiridol in Combination with Docetaxel in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2004, 10, 5038–5047. [Google Scholar] [CrossRef]
- Shiradkar, M.R.; Akula, K.C.; Dasari, V.; Baru, V.; Chiningiri, B.; Gandhi, S.; Kaur, R. Clubbed Thiazoles by MAOS: A Novel Approach to Cyclin-Dependent Kinase 5/P25 Inhibitors as a Potential Treatment for Alzheimer’s Disease. Bioorg. Med. Chem. 2007, 15, 2601–2610. [Google Scholar] [CrossRef]
- Lenjisa, J.L.; Tadesse, S.; Khair, N.Z.; Kumarasiri, M.; Yu, M.; Albrecht, H.; Milne, R.; Wang, S. CDK5 in Oncology: Recent Advances and Future Prospects. Future Med. Chem. 2017, 9, 1939–1962. [Google Scholar] [CrossRef]
- Tekman, S.; Öner, N. A Pentose Identified in the Carbohydrate Group of Spongin. Nature 1963, 200, 77–78. [Google Scholar] [CrossRef]
- Lyu, C.; Chen, T.; Qiang, B.; Liu, N.; Wang, H.; Zhang, L.; Liu, Z. CMNPD: A Comprehensive Marine Natural Products Database towards Facilitating Drug Discovery from the Ocean. Nucleic Acids Res. 2021, 49, D509–D515. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Investigation of Marine-Derived Natural Products as Raf Kinase Inhibitory Protein (Rkip)-Binding Ligands. Mar. Drugs 2021, 19, 581. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Lee, G.; Rampogu, S.; Hong, J.C.; Lee, K.W. Marine-Derived Natural Products as ATP-Competitive MTOR Kinase Inhibitors for Cancer Therapeutics. Pharmaceuticals 2021, 14, 282. [Google Scholar] [CrossRef]
- Veselovsky, A.; Ivanov, A. Strategy of Computer-Aided Drug Design. Curr. Drug Target Infectious Disord. 2003, 3, 33–40. [Google Scholar] [CrossRef]
- Saur, I.M.L.; Panstruga, R.; Schulze-Lefert, P. NOD-like Receptor-Mediated Plant Immunity: From Structure to Cell Death. Nat. Rev. Immunol. 2021, 21, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, T.H.; Abdelmoniem, N.; Mukhtar, R.M.; Alqhtani, A.T.; Alalawi, A.L.; Alawaji, R.; Althubyani, M.S.; Mohamed, S.G.A.; Mohamed, G.A.; Ibrahim, S.R.M.; et al. Molecular Docking and Molecular Dynamics Studies Reveal the Anticancer Potential of Medicinal-Plant-Derived Lignans as MDM2-P53 Interaction Inhibitors. Molecules 2023, 28, 6665. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Dhasmana, A.; Raza, S.; Jahan, R.; Lohani, M.; Arif, J.M. High-Throughput Virtual Screening (HTVS) of Natural Compounds and Exploration of Their Biomolecular Mechanisms: An In Silico Approach. In New Look to Phytomedicine: Advancements in Herbal Products as Novel Drug Leads; Elsevier: Amsterdam, The Netherlands, 2018; pp. 523–548. ISBN 9780128146200. [Google Scholar]
- Repasky, M.P.; Murphy, R.B.; Banks, J.L.; Greenwood, J.R.; Tubert-Brohman, I.; Bhat, S.; Friesner, R.A. Docking Performance of the Glide Program as Evaluated on the Astex and DUD Datasets: A Complete Set of Glide SP Results and Selected Results for a New Scoring Function Integrating WaterMap and Glide. J. Comput. Aided. Mol. Des. 2012, 26, 787–799. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Beard, H.; Cholleti, A.; Pearlman, D.; Sherman, W.; Loving, K.A. Applying Physics-Based Scoring to Calculate Free Energies of Binding for Single Amino Acid Mutations in Protein-Protein Complexes. PLoS ONE 2013, 8, e82849. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of Clinical Drug Development Fails and How to Improve It? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Singh, S.; Bani Baker, Q.; Singh, D.B. Molecular Docking and Molecular Dynamics Simulation. In Bioinformatics: Methods and Applications; Elsevier: Amsterdam, The Netherlands, 2021; pp. 291–304. ISBN 9780323897754. [Google Scholar]
- Mukhtar, R.M.; Abdelmoniem, N.; Elrufaie, H.A.; Edris, A.; Ghaboosh, H.; Mahgoub, M.A.; Garelnabi, E.A.E.; Osman, W.; Sherif, A.E.; Ashour, A.; et al. Unlocking the Potential of Approved Drugs for the Allosteric Inhibition of Tropomyosin-Receptor Kinase A Using Molecular Docking and Molecular Dynamics Studies. Front. Chem. 2023, 11, 1205724. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2012, 7, 146–157. [Google Scholar] [CrossRef]
- Romano, T. Kroemer Structure-Based Drug Design: Docking and Scoring. Curr. Protein Pept. Sci. 2007, 8, 312–328. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 5. Improved Docking Performance Using High Solute Dielectric Constant MM/GBSA and MM/PBSA Rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Ahinko, M.; Niinivehmas, S.; Jokinen, E.; Pentikäinen, O.T. Suitability of MMGBSA for the Selection of Correct Ligand Binding Modes from Docking Results. Chem. Biol. Drug Des. 2019, 93, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the Molecular Mechanics/Poisson Boltzmann Surface Area and Molecular Mechanics/Generalized Born Surface Area Methods. II. the Accuracy of Ranking Poses Generated from Docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, S.; Liu, D.; Proksch, P.; Lin, W. Inhibitory Effects of Polyphenols toward HCV from the Mangrove Plant Excoecaria agallocha L. Bioorganic Med. Chem. Lett. 2012, 22, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Utkina, N.K.; Makarchenko, A.E.; Denisenko, V.A. Zyzzyanones B-D, Dipyrroloquinones from the Marine Sponge Zyzzya Fuliginosa. J. Nat. Prod. 2005, 68, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Nijampatnam, B.; Dutta, S.; Velu, S.E. Recent Advances in Isolation, Synthesis, and Evaluation of Bioactivities of Bispyrroloquinone Alkaloids of Marine Origin. Chin. J. Nat. Med. 2015, 13, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Plisson, F.; Huang, X.C.; Zhang, H.; Khalil, Z.; Capon, R.J. Lamellarins as Inhibitors of P-Glycoprotein-Mediated Multidrug Resistance in a Human Colon Cancer Cell Line. Chem. An Asian J. 2012, 7, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.N.; Zhang, H.J.; Li, J.Q.; Ding, W.J.; Ma, Z.J. Bioactive Indolocarbazoles from the Marine-Derived Streptomyces Sp. DT-A61. J. Nat. Prod. 2018, 81, 949–956. [Google Scholar] [CrossRef]
- Xiao, F.; Li, H.; Xu, M.; Li, T.; Wang, J.; Sun, C.; Hong, K.; Li, W. Staurosporine Derivatives Generated by Pathway Engineering in a Heterologous Host and Their Cytotoxic Selectivity. J. Nat. Prod. 2018, 81, 1745–1751. [Google Scholar] [CrossRef]
- Rowley, D.C.; Hansen, M.S.T.; Rhodes, D.; Sotriffer, C.A.; Ni, H.; McCammon, J.A.; Bushman, F.D.; Fenical, W. Thalassiolins A-C: New Marine-Derived Inhibitors of HIV CDNA Integrase. Bioorganic Med. Chem. 2002, 10, 3619–3625. [Google Scholar] [CrossRef]
- Pereira, A.; Vottero, E.; Roberge, M.; Mauk, A.G.; Andersen, R.J. Indoleamine 2,3-Dioxygenase Inhibitors from the Northeastern Pacific Marine Hydroid Garveia Annulata. J. Nat. Prod. 2006, 69, 1496–1499. [Google Scholar] [CrossRef]
- Alonso, E.; Alvariño, R.; Leirós, M.; Tabudravu, J.N.; Feussner, K.; Dam, M.A.; Rateb, M.E.; Jaspars, M.; Botana, L.M. Evaluation of the Antioxidant Activity of the Marine Pyrroloiminoquinone Makaluvamines. Mar. Drugs 2016, 14, 197. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Duffy, E.M. Prediction of Drug Solubility from Structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating Molecular Docking and Molecular Dynamics Simulations. In Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 2053, pp. 13–34. ISBN 9781493997527. [Google Scholar]
- Shukla, R.; Tripathi, T. Molecular Dynamics Simulation of Protein and Protein–Ligand Complexes. In Computer-Aided Drug Design; Springer: Singapore, 2020; pp. 133–161. ISBN 9789811568152. [Google Scholar]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, T.H.; Ibraheem, W.; Abdelrahman, M.; Osman, W.; Sherif, A.E.; Ashour, A.; Ibrahim, S.R.M.; Ghazawi, K.F.; Miski, S.F.; Almadani, S.A.; et al. Exploring the Potential of Approved Drugs for Triple-Negative Breast Cancer Treatment by Targeting Casein Kinase 2: Insights from Computational Studies. PLoS ONE 2023, 18, e0289887. [Google Scholar] [CrossRef]








| Compound | Hydrogen Bonds | Hydrophobic Interactions | Other Interactions | |
|---|---|---|---|---|
| Residue | Distance (Å) | |||
| Excoecariphenol B | Gln51 Cys83 Asp144 | 1.93 1.70 2.39 | Ile10, Tyr15, Val18, Ala31, Val64, Phe80, Phe82, Cys83, and Leu133 | - |
| Excoecariphenol A | Gln51 Cys83 | 1.83 1.68 | Ile10, Ala31, Val64, Phe80, Phe82, Cys83, and Leu133 | - |
| Zyzzyanone B | Gln51 Glu81 Asp86 Cys83 | 1.85 2.25 2.11 1.94, 1.67 | Ile10, Tyr15, Val18, Ala31, Phe80, Phe82, Cys83, and Leu133 | salt bridge-Asp86 Pi-Pi stacking-Phe80 |
| Reference | Lys33 Gln51 Cys83 Asp86 Asp144 | 2.76 1.87 1.97, 1.84 2.08 1.98, 2.39 | Ile10, Tyr15, Val18, Ala31, Val64, Phe80, Phe82, Cys83, and Leu133 | salt bridge-Asp144 Pi-cation-Tyr15 |
| HBD a | HBA b | QPlog Po/w c | QPlog S d | QPlog HERG e | QPP Caco f | QPlog BB g | Mwt h | Rule Of Five i | |
|---|---|---|---|---|---|---|---|---|---|
| Excoecariphenol B | 10 | 15.2 | −2.033 | −2.489 | −5.025 | 1.377 | −4.213 | 500.473 | 3 |
| Excoecariphenol A | 9 | 14.45 | −1.406 | −2.741 | −5.305 | 4.115 | −3.69 | 484.474 | 2 |
| Zyzzyanone B | 4 | 6.25 | 1.076 | −3.043 | −6.275 | 22.892 | −1.397 | 335.362 | 0 |
| Standard values | ≤5 | ≤10 | −2.0–6.5 | −6.5˗0.5 | Below −5 | >25 poor <500 great | −3–1.2 | >500 | 0˗4 |
| Compound | Oral Toxicity | Organ Toxicity | Toxicity Endpoints Prediction | ||||
|---|---|---|---|---|---|---|---|
| Toxicity Class | Predicted LD50 (mg/kg) | Hepatotoxicity | Mutagenicity | Cyto Toxicity | Carcinogenicity | Immunotoxicity | |
| Excoecariphenol B | 5 | 2500 | inactive | inactive | inactive | inactive | inactive |
| Excoecariphenol A | 5 | 2500 | inactive | inactive | inactive | inactive | inactive |
| Zyzzyanone B | 4 | 1000 | inactive | inactive | inactive | inactive | inactive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoaib, T.H.; Almogaddam, M.A.; Andijani, Y.S.; Saib, S.A.; Almaghrabi, N.M.; Elyas, A.F.; Azzouni, R.Y.; Awad, E.A.; Mohamed, S.G.A.; Mohamed, G.A.; et al. Marine-Derived Compounds for CDK5 Inhibition in Cancer: Integrating Multi-Stage Virtual Screening, MM/GBSA Analysis and Molecular Dynamics Investigations. Metabolites 2023, 13, 1090. https://doi.org/10.3390/metabo13101090
Shoaib TH, Almogaddam MA, Andijani YS, Saib SA, Almaghrabi NM, Elyas AF, Azzouni RY, Awad EA, Mohamed SGA, Mohamed GA, et al. Marine-Derived Compounds for CDK5 Inhibition in Cancer: Integrating Multi-Stage Virtual Screening, MM/GBSA Analysis and Molecular Dynamics Investigations. Metabolites. 2023; 13(10):1090. https://doi.org/10.3390/metabo13101090
Chicago/Turabian StyleShoaib, Tagyedeen H., Mohammed A. Almogaddam, Yusra Saleh Andijani, Samaher Ahmad Saib, Najwa Mahmoud Almaghrabi, Abdulaziz Fahad Elyas, Rahmah Yasin Azzouni, Ehda Ahmad Awad, Shaimaa G. A. Mohamed, Gamal A. Mohamed, and et al. 2023. "Marine-Derived Compounds for CDK5 Inhibition in Cancer: Integrating Multi-Stage Virtual Screening, MM/GBSA Analysis and Molecular Dynamics Investigations" Metabolites 13, no. 10: 1090. https://doi.org/10.3390/metabo13101090
APA StyleShoaib, T. H., Almogaddam, M. A., Andijani, Y. S., Saib, S. A., Almaghrabi, N. M., Elyas, A. F., Azzouni, R. Y., Awad, E. A., Mohamed, S. G. A., Mohamed, G. A., Ibrahim, S. R. M., Hussein, H. G. A., Osman, W., Ashour, A., Sherif, A. E., & Alzain, A. A. (2023). Marine-Derived Compounds for CDK5 Inhibition in Cancer: Integrating Multi-Stage Virtual Screening, MM/GBSA Analysis and Molecular Dynamics Investigations. Metabolites, 13(10), 1090. https://doi.org/10.3390/metabo13101090