Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS)

Abstract
:1. Introduction
2. Results
2.1. Extraction of PIPs

2.2. Mass Spectrometric Characterization of DAGs and PIPs


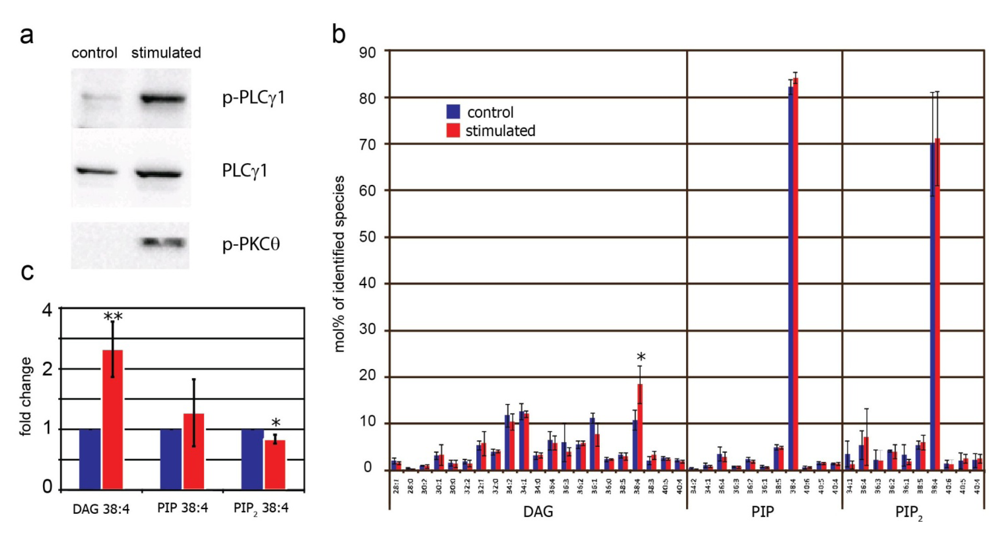
2.3. Identification and Quantification of DAG, PIP and PIP2 in Human T Cells


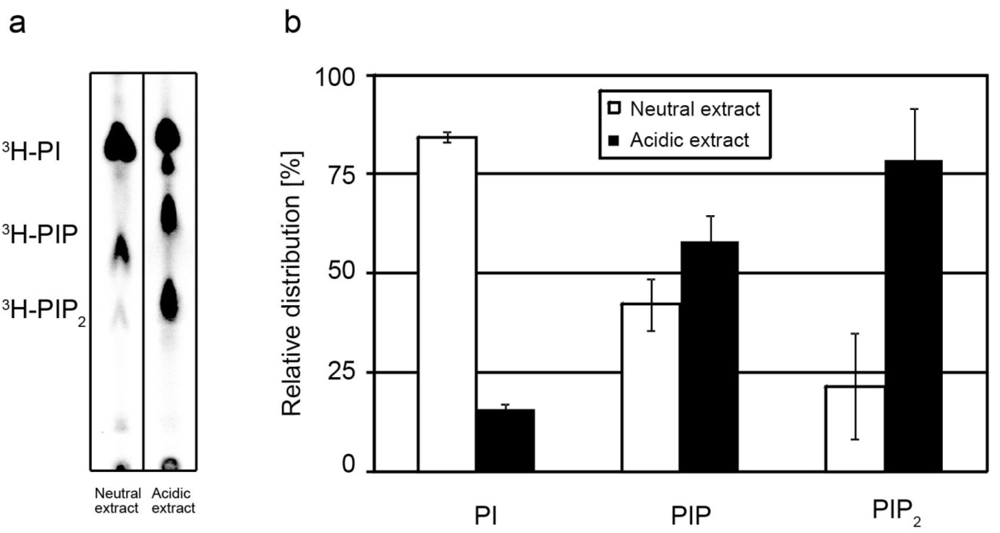{kind=link}
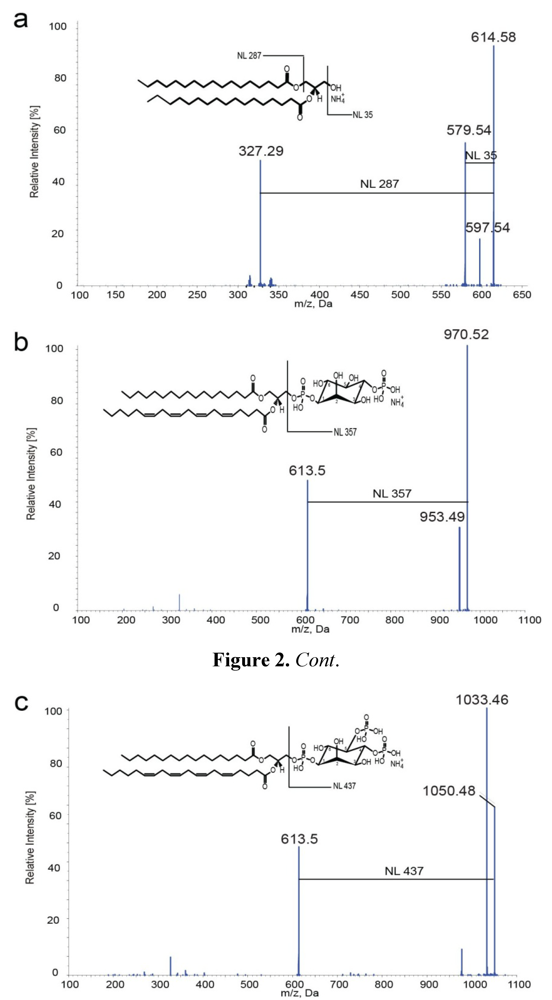{kind=link}
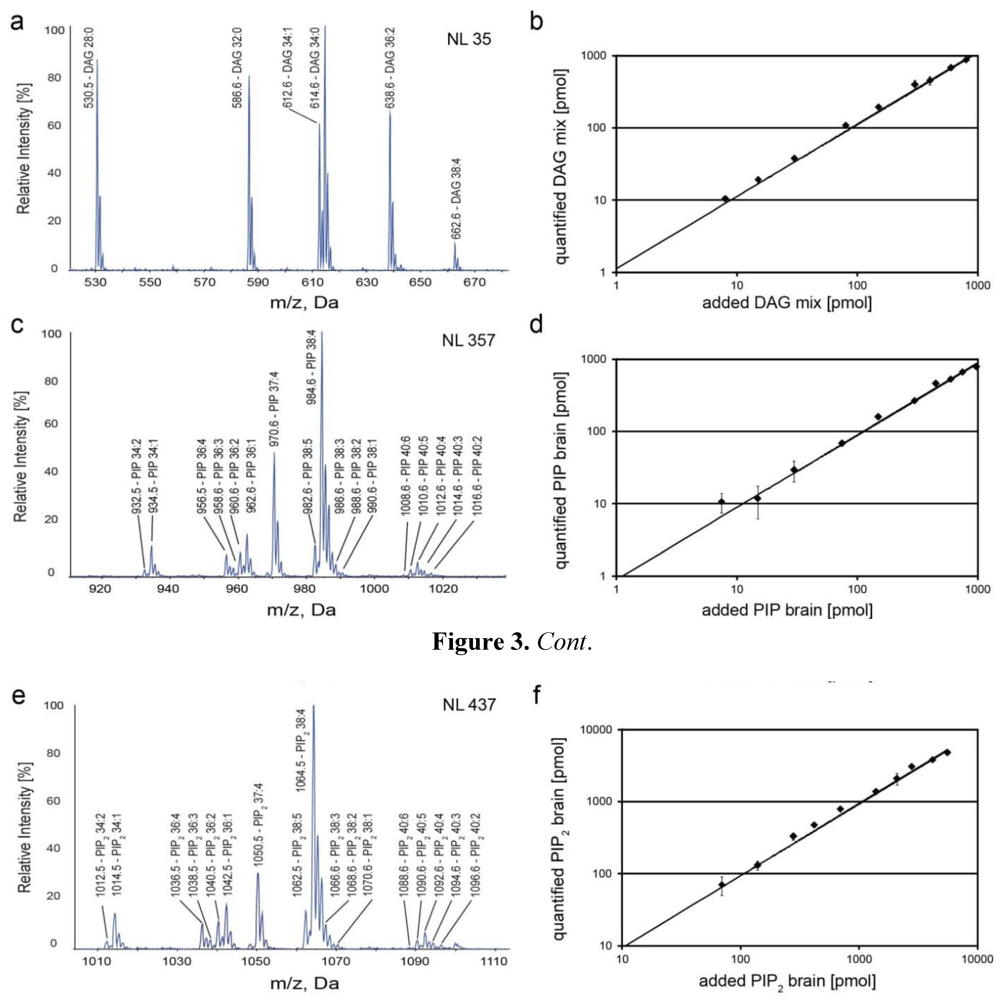{kind=link}
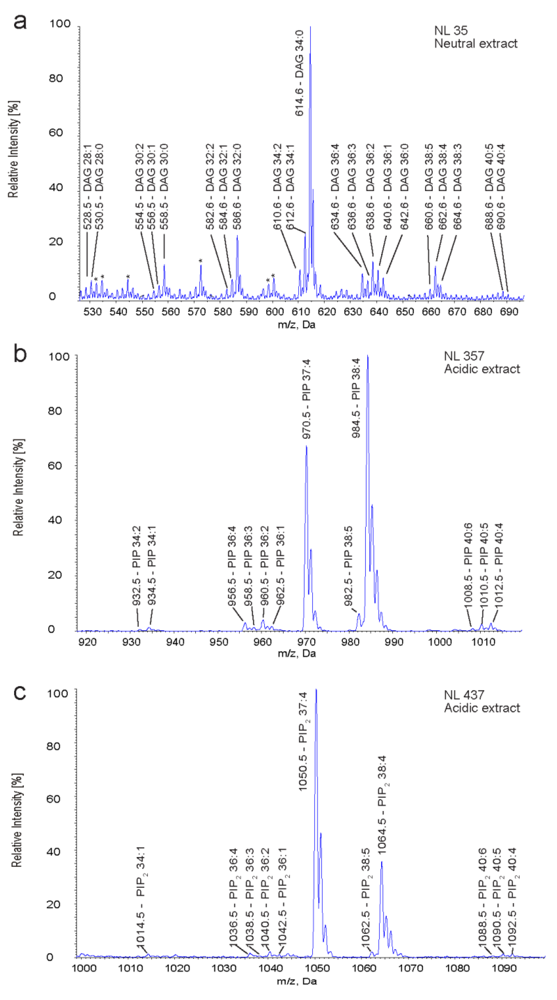{kind=link}
{kind=link}
| Species | DAG | PIP | PIP2 | |||
|---|---|---|---|---|---|---|
| m/z | mol% | m/z | mol% | m/z | mol% | |
| 28:1 | 528.5 | 2.1 ± 0.6 | - | - | - | - |
| 28:0 | 530.5 | 0.5 ± 0.2 | - | - | - | - |
| 30:2 | 554.5 | 1.0 ± 0.1 | - | - | - | - |
| 30:1 | 556.5 | 3.1 ± 0.8 | - | - | - | - |
| 30:0 | 558.5 | 1.6 ± 0.6 | - | - | - | - |
| 32:2 | 582.6 | 1.9 ± 0.5 | - | - | - | - |
| 32:1 | 584.6 | 5.4 ± 0.9 | - | - | - | - |
| 32:0 | 586.6 | 3.9 ± 0.6 | - | - | - | - |
| 34:2 | 610.6 | 11.8 ± 2.5 | 932.5 | 0.5 ± 0.2 | - | - |
| 34:1 | 612.6 | 12.6 ± 1.8 | 934.5 | 1.1 ± 0.4 | 1014.5 | 3.6 ± 2.8 |
| 34:0 | 614.6 | 3.2 ± 0.8 | - | - | - | - |
| 36:4 | 634.6 | 6.4 ± 1.9 | 956.5 | 3.6 ± 1.5 | 1036.5 | 5.5 ± 3.1 |
| 36:3 | 636.6 | 6.0 ± 4.1 | 958.5 | 0.8 ± 0.1 | 1038.5 | 2.2 ± 2.2 |
| 36:2 | 638.6 | 5.5 ± 0.8 | 960.5 | 2.4 ± 0.5 | 1040.5 | 4.2 ± 0.3 |
| 36:1 | 640.6 | 11.2 ± 1.1 | 962.5 | 0.9 ± 0.2 | 1042.5 | 3.3 ± 2.3 |
| 36:0 | 642.6 | 2.4 ± 0.4 | - | - | - | - |
| 38:5 | 660.6 | 3.3 ± 0.6 | 982.5 | 5.0 ± 0.6 | 1062.5 | 5.5 ± 0.9 |
| 38:4 | 662.6 | 10.8 ± 2.2 | 984.5 | 82.2 ± 1.5 | 1064.5 | 70.0 ± 11.1 |
| 38:3 | 664.6 | 2.1 ± 0.9 | - | - | - | - |
| 40:6 | - | - | 1008.5 | 0.6 ± 0.3 | 1088.5 | 1.4 ± 0.8 |
| 40:5 | 688.6 | 2.7 ± 0.4 | 1010.5 | 1.6 ± 0.3 | 1090.5 | 2.1 ± 1.8 |
| 40:4 | 690.6 | 2.3 ± 0.3 | 1012.5 | 1.5 ± 0.2 | 1092.5 | 2.2 ± 1.5 |
2.4. Effect of TCR Stimulation on DAG, PIP and PIP2 in Human T Cells

3. Discussion
4. Experimental Section
| QStar® Elite | QTrap® 5500 | |
|---|---|---|
| Curtain Gas | 10 | 10 |
| CAD Gas | 2 | 5 |
| Operating pressure (torr) | 2.5 × 10−5 | 1.6 × 10−5 |
| Interface heater temperature IHT (°C) | 40 | 40 |
| Declustering potential (DP) | 40 | 100 |
| Focusing potential (FP) | 200 | - |
| Declustering potential 2 (DP2) | 10 | - |
| Entrance potential (EP) | - | 7 |
| Collision cell exit potential (CXP) | - | 19 |
| Detector (CEM) | 2,500 | 2,100 |
| Quadrupole resolution | Unit (Q1) | Unit (Q1 and Q3) |
| MAG-H2O | 14:1 | 14:0 | 16:2 | 16:1 | 16:0 | 17:0 | 18:3 | 18:2 | 18:1 |
| m/z | 283.2 | 285.2 | 309.2 | 311.3 | 313.3 | 327.3 | 335.3 | 337.3 | 339.3 |
| MAG-H2O | 18:0 | 20:5 | 20:4 | 20:3 | 20:2 | 20:1 | 20:0 | 22:6 | 22:5 |
| m/z | 341.3 | 359.3 | 361.3 | 363.3 | 365.3 | 367.3 | 369.3 | 385.3 | 387.3 |
5. Conclusions
Supplementary Materials
Acknowledgments
Conflict of Interest
References
- Sasaki, T.; Takasuga, S.; Sasaki, J.; Kofuji, S.; Eguchi, S.; Yamazaki, M.; Suzuki, A. Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 2009, 48, 307–343. [Google Scholar] [CrossRef]
- Tolias, K.F.; Cantley, L.C. Pathways for phosphoinositide synthesis. Chem. Phys. Lipids 1999, 98, 69–77. [Google Scholar] [CrossRef]
- Blero, D.; Payrastre, B.; Schurmans, S.; Erneux, C. Phosphoinositide phosphatases in a network of signalling reactions. Pflugers Arch. 2007, 455, 31–44. [Google Scholar] [CrossRef]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef]
- Downes, C.P.; Gray, A.; Fairservice, A.; Safrany, S.T.; Batty, I.H.; Fleming, I. The regulation of membrane to cytosol partitioning of signalling proteins by phosphoinositides and their soluble headgroups. Biochem. Soc. Trans. 2005, 33, 1303–1307. [Google Scholar] [CrossRef]
- De Matteis, M.A.; Di Campli, A.; Godi, A. The role of the phosphoinositides at the Golgi complex. Biochim. Biophys. Acta 1744, 396–405. [Google Scholar]
- Rojo, J.M.; Bello, R.; Portoles, P. T-cell receptor. Adv. Exp. Med. Biol. 2008, 640, 1–11. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Huang, Y.H.; Sauer, K. Lipid signaling in T-cell development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a002428. [Google Scholar] [CrossRef]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef]
- Hama, H.; Torabinejad, J.; Prestwich, G.D.; DeWald, D.B. Measurement and immunofluorescence of cellular phosphoinositides. Methods Mol. Biol. 2004, 284, 243–258. [Google Scholar]
- Bird, I.M. Analysis of cellular phosphoinositides and phosphoinositols by extraction and simple analytical procedures. Methods Mol. Biol. 1994, 27, 227–248. [Google Scholar]
- Cooke, F.T. Measurement of polyphosphoinositides in cultured mammalian cells. Methods Mol. Biol. 2009, 462, 43–58. [Google Scholar] [CrossRef]
- Bielawska, A.; Perry, D.K.; Hannun, Y.A. Determination of ceramides and diglycerides by the diglyceride kinase assay. Anal. Biochem. 2001, 298, 141–150. [Google Scholar]
- Falardeau, P.; Robillard, M.; Hui, R. Quantification of diacylglycerols by capillary gas chromatography-negative ion chemical ionization-mass spectrometry. Anal. Biochem. 1993, 208, 311–316. [Google Scholar]
- Hubbard, W.C.; Hundley, T.R.; Oriente, A.; MacGlashan, D.W., Jr. Quantitation of 1-stearoyl-2-arachidonoyl-sn-3-glycerol in human basophils via gas chromatography-negative ion chemical ionization mass spectrometry. Anal. Biochem. 1996, 236, 309–321. [Google Scholar]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar]
- Blanksby, S.J.; Mitchell, T.W. Advances in mass spectrometry for lipidomics. Annu. Rev. Anal. Chem. (Palo Alto Calif) 2010, 3, 433–465. [Google Scholar] [CrossRef]
- Han, X.; Gross, R.W. Electrospray ionization mass spectroscopic analysis of human erythrocyte plasma membrane phospholipids. Proc. Natl. Acad. Sci. USA 1994, 91, 10635–10639. [Google Scholar] [CrossRef]
- Kerwin, J.L.; Tuininga, A.R.; Ericsson, L.H. Identification of molecular species of glycerophospholipids and sphingomyelin using electrospray mass spectrometry. J. Lipid Res. 1994, 35, 1102–1114. [Google Scholar]
- Hsu, F.F.; Turk, J. Structural determination of glycosphingolipids as lithiated adducts by electrospray ionization mass spectrometry using low-energy collisional-activated dissociation on a triple stage quadrupole instrument. J. Am. Soc. Mass Spectrom. 2001, 12, 61–79. [Google Scholar] [CrossRef]
- Han, X.; Gross, R.W. Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: A bridge to lipidomics. J. Lipid Res. 2003, 44, 1071–1079. [Google Scholar] [CrossRef]
- Ivanova, P.T.; Milne, S.B.; Byrne, M.O.; Xiang, Y.; Brown, H.A. Glycerophospholipid identification and quantitation by electrospray ionization mass spectrometry. Methods Enzymol 2007, 432, 21–57. [Google Scholar]
- Kainu, V.; Hermansson, M.; Somerharju, P. Electrospray ionization mass spectrometry and exogenous heavy isotope-labeled lipid species provide detailed information on aminophospholipid acyl chain remodeling. J. Biol. Chem. 2008, 283, 3676–3687. [Google Scholar]
- Scherer, M.; Bottcher, A.; Liebisch, G. Lipid profiling of lipoproteins by electrospray ionization tandem mass spectrometry. Biochim. Biophys. Acta 1811, 918–924. [Google Scholar]
- Houjou, T.; Yamatani, K.; Imagawa, M.; Shimizu, T.; Taguchi, R. A shotgun tandem mass spectrometric analysis of phospholipids with normal-phase and/or reverse-phase liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 654–666. [Google Scholar]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar]
- Han, X.; Gross, R.W. Shotgun lipidomics: Electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom. Rev. 2005, 24, 367–412. [Google Scholar] [CrossRef]
- Stahlman, M.; Ejsing, C.S.; Tarasov, K.; Perman, J.; Boren, J.; Ekroos, K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2664–2672. [Google Scholar] [CrossRef]
- Han, X.; Yang, K.; Yang, J.; Cheng, H.; Gross, R.W. Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J. Lipid Res. 2006, 47, 864–879. [Google Scholar] [CrossRef]
- Sampaio, J.L.; Gerl, M.J.; Klose, C.; Ejsing, C.S.; Beug, H.; Simons, K.; Shevchenko, A. Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. USA 2006, 108, 1903–1907. [Google Scholar]
- Brugger, B.; Erben, G.; Sandhoff, R.; Wieland, F.T.; Lehmann, W.D. Quantitative analysis of biological membrane lipids at the low picomole level by nano-electrospray ionization tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 1997, 94, 2339–2344. [Google Scholar]
- Ejsing, C.S.; Duchoslav, E.; Sampaio, J.; Simons, K.; Bonner, R.; Thiele, C.; Ekroos, K.; Shevchenko, A. Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal. Chem. 2006, 78, 6202–6214. [Google Scholar]
- Murphy, R.C.; James, P.F.; McAnoy, A.M.; Krank, J.; Duchoslav, E.; Barkley, R.M. Detection of the abundance of diacylglycerol and triacylglycerol molecular species in cells using neutral loss mass spectrometry. Anal. Biochem. 2007, 366, 59–70. [Google Scholar]
- Schwudke, D.; Oegema, J.; Burton, L.; Entchev, E.; Hannich, J.T.; Ejsing, C.S.; Kurzchalia, T.; Shevchenko, A. Lipid profiling by multiple precursor and neutral loss scanning driven by the data-dependent acquisition. Anal. Chem. 2006, 78, 585–595. [Google Scholar] [CrossRef]
- Ekroos, K.; Chernushevich, I.V.; Simons, K.; Shevchenko, A. Quantitative profiling of phospholipids by multiple precursor ion scanning on a hybrid quadrupole time-of-flight mass spectrometer. Anal. Chem. 2002, 74, 941–949. [Google Scholar] [CrossRef]
- Wakelam, M.J.; Clark, J. Methods for analyzing phosphoinositides using mass spectrometry. Biochim. Biophys. Acta 1811, 758–762. [Google Scholar]
- Milne, S.B.; Ivanova, P.T.; DeCamp, D.; Hsueh, R.C.; Brown, H.A. A targeted mass spectrometric analysis of phosphatidylinositol phosphate species. J. Lipid Res. 2005, 46, 1796–1802. [Google Scholar] [CrossRef]
- Hsu, F.F.; Turk, J. Characterization of phosphatidylinositol, phosphatidylinositol-4-phosphate, and phosphatidylinositol-4,5-bisphosphate by electrospray ionization tandem mass spectrometry: A mechanistic study. J. Am. Soc. Mass Spectrom. 2000, 11, 986–999. [Google Scholar] [CrossRef]
- Pettitt, T.R.; Dove, S.K.; Lubben, A.; Calaminus, S.D.; Wakelam, M.J. Analysis of intact phosphoinositides in biological samples. J. Lipid Res. 2006, 47, 1588–1596. [Google Scholar] [CrossRef]
- Ogiso, H.; Taguchi, R. Reversed-phase LC/MS method for polyphosphoinositide analyses: changes in molecular species levels during epidermal growth factor activation in A431 cells. Anal. Chem. 2008, 80, 9226–9232. [Google Scholar] [CrossRef]
- Clark, J.; Anderson, K.E.; Juvin, V.; Smith, T.S.; Karpe, F.; Wakelam, M.J.; Stephens, L.R.; Hawkins, P.T. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat. Methods 2011, 8, 267–272. [Google Scholar]
- Wenk, M.R.; Lucast, L.; Di Paolo, G.; Romanelli, A.J.; Suchy, S.F.; Nussbaum, R.L.; Cline, G.W.; Shulman, G.I.; McMurray, W.; De Camilli, P. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat. Biotechnol. 2003, 21, 813–817. [Google Scholar] [CrossRef]
- Callender, H.L.; Forrester, J.S.; Ivanova, P.; Preininger, A.; Milne, S.; Brown, H.A. Quantification of diacylglycerol species from cellular extracts by electrospray ionization mass spectrometry using a linear regression algorithm. Anal. Chem. 2007, 79, 263–272. [Google Scholar] [CrossRef]
- Li, Y.L.; Su, X.; Stahl, P.D.; Gross, M.L. Quantification of diacylglycerol molecular species in biological samples by electrospray ionization mass spectrometry after one-step derivatization. Anal. Chem. 2007, 79, 1569–1574. [Google Scholar]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Dawson, R.M.; Eichberg, J. Diphosphoinositide and triphosphoinositide in animal tissues. Extraction, estimation and changes post mortem. Biochem. J. 1965, 96, 634–643. [Google Scholar]
- Michell, R.H.; Hawthorne, J.N.; Coleman, R.; Karnovsky, M.L. Extraction of polyphosphoinositides with neutral and acidified solvents. A comparison of guinea-pig brain and liver, and measurements of rat liver inositol compounds which are resistant to extraction. Biochim. Biophys. Acta 1970, 210, 86–91. [Google Scholar]
- Hama, H.; Takemoto, J.Y.; DeWald, D.B. Analysis of phosphoinositides in protein trafficking. Methods 2000, 20, 465–473. [Google Scholar] [CrossRef]
- Vickers, J.D. Extraction of polyphosphoinositides from platelets: Comparison of a two-step procedure with a common single-step extraction procedure. Anal. Biochem. 1995, 224, 449–451. [Google Scholar] [CrossRef]
- Gray, A.; Olsson, H.; Batty, I.H.; Priganica, L.; Peter Downes, C. Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal. Biochem. 2003, 313, 234–245. [Google Scholar] [CrossRef]
- Han, X.; Yang, J.; Cheng, H.; Ye, H.; Gross, R.W. Toward fingerprinting cellular lipidomes directly from biological samples by two-dimensional electrospray ionization mass spectrometry. Anal. Biochem. 2004, 330, 317–331. [Google Scholar]
- Gottfried, E.L. Lipids of human leukocytes: relation to celltype. J. Lipid Res. 1967, 8, 321–327. [Google Scholar]
- Zaru, R.; Berrie, C.P.; Iurisci, C.; Corda, D.; Valitutti, S. CD28 co-stimulates TCR/CD3-induced phosphoinositide turnover in human T lymphocytes. Eur. J. Immunol. 2001, 31, 2438–2447. [Google Scholar] [CrossRef]
- Mallo, G.V.; Espina, M.; Smith, A.C.; Terebiznik, M.R.; Aleman, A.; Finlay, B.B.; Rameh, L.E.; Grinstein, S.; Brumell, J.H. SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J. Cell Biol. 2008, 182, 741–752. [Google Scholar] [CrossRef]
- Zemski Berry, K.A.; Murphy, R.C. Electrospray ionization tandem mass spectrometry of glycerophosphoethanolamine plasmalogen phospholipids. J. Am. Soc. Mass Spectrom. 2004, 15, 1499–1508. [Google Scholar] [CrossRef]
- Mor, A.; Philips, M.R. Compartmentalized Ras/MAPK signaling. Annu. Rev. Immunol. 2006, 24, 771–800. [Google Scholar] [CrossRef]
- Mor, A.; Campi, G.; Du, G.; Zheng, Y.; Foster, D.A.; Dustin, M.L.; Philips, M.R. The lymphocyte function-associated antigen-1 receptor costimulates plasma membrane Ras via phospholipase D2. Nat. Cell Biol. 2007, 9, 713–719. [Google Scholar] [CrossRef]
- Huse, M. The T-cell-receptor signaling network. J. Cell Sci. 2009, 122, 1269–1273. [Google Scholar] [CrossRef]
- Rouser, G.; Fkeischer, S.; Yamamoto, A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 1970, 5, 494–496. [Google Scholar] [CrossRef]
- Brugger, B.; Graham, C.; Leibrecht, I.; Mombelli, E.; Jen, A.; Wieland, F.; Morris, R. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J. Biol. Chem. 2004, 279, 7530–7536. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Haag, M.; Schmidt, A.; Sachsenheimer, T.; Brügger, B. Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS). Metabolites 2012, 2, 57-76. https://doi.org/10.3390/metabo2010057
Haag M, Schmidt A, Sachsenheimer T, Brügger B. Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS). Metabolites. 2012; 2(1):57-76. https://doi.org/10.3390/metabo2010057
Chicago/Turabian StyleHaag, Mathias, Angelika Schmidt, Timo Sachsenheimer, and Britta Brügger. 2012. "Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS)" Metabolites 2, no. 1: 57-76. https://doi.org/10.3390/metabo2010057
APA StyleHaag, M., Schmidt, A., Sachsenheimer, T., & Brügger, B. (2012). Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS). Metabolites, 2(1), 57-76. https://doi.org/10.3390/metabo2010057