The Complex Role of Branched Chain Amino Acids in Diabetes and Cancer

Abstract
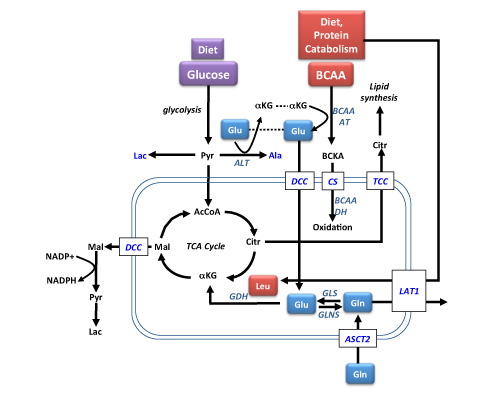
:
{kind=link}
{kind=link}
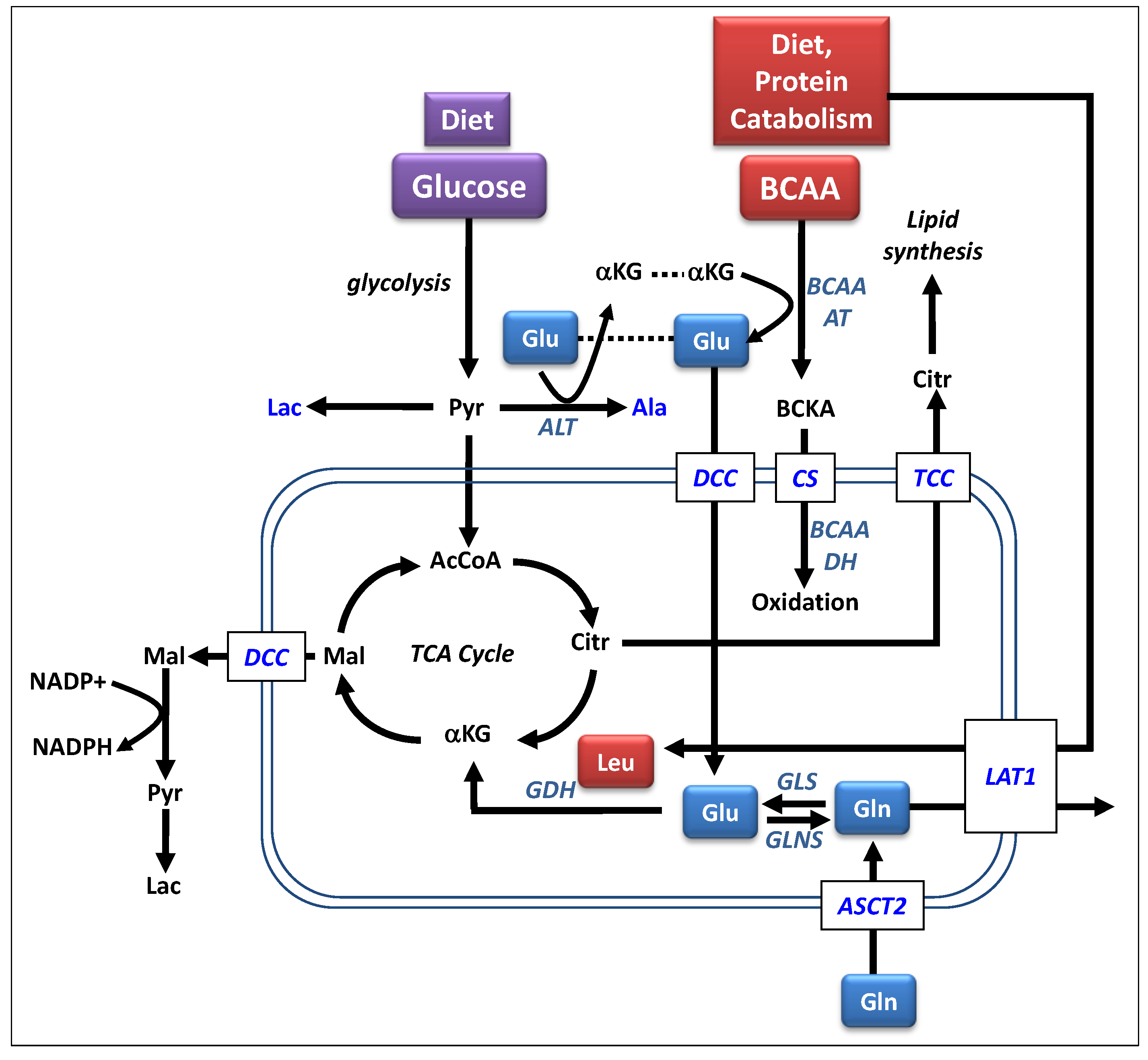{kind=link}
{kind=link}
1. Introduction
2. BCAAs in Bioenergetics and Protein Synthesis

3. The Role of BCAAs in Diabetes
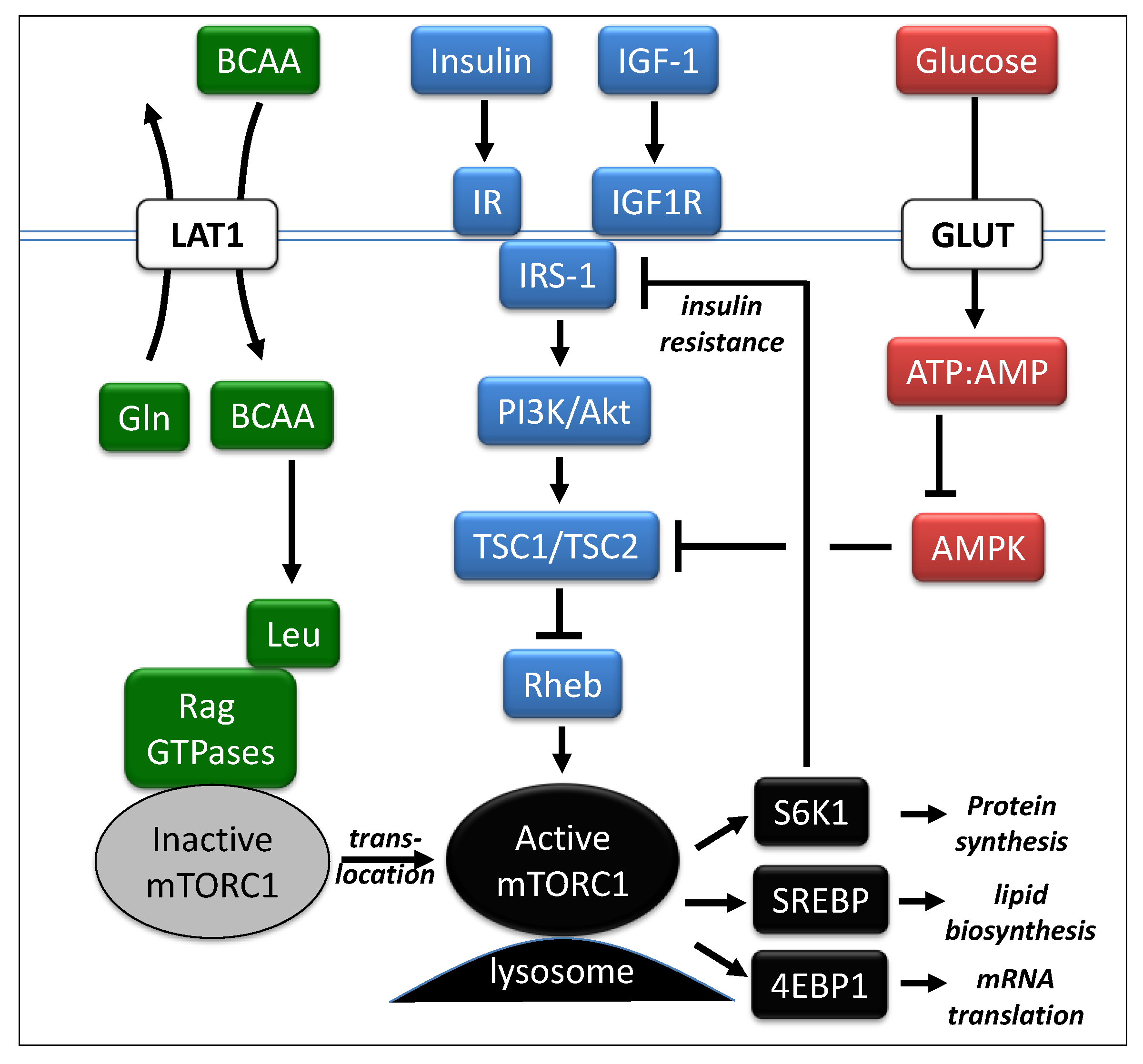
4. Amino Acids, Glucose and the mTORC1 Pathway

5. BCAAs in Cancer
6. BCAAs in Cachexia
7. Therapeutic Use of BCAA Supplementation
8. Conclusions
Disclaimer
Conflicts of Interest
References
- Gallagher, E.J.; Leroith, D. Epidemiology and molecular mechanisms tying obesity, diabetes and the metabolic syndrome with cancer. Diabetes Care 2013, 36, S233–S239. [Google Scholar] [CrossRef]
- Johnson, J.A.; Carstensen, B.; Witte, D.; Bowker, S.L.; Lipscombe, L.; Renehan, A.G. Diabetes and cancer (1): Evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 2012, 55, 1607–1618. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Felig, P.; Marliss, E.B.; Cahill, G.F. Plasma amino acid levels and insulin secretion in obesity. N. Engl. J. Med. 1969, 281, 811–816. [Google Scholar] [CrossRef]
- Felig, P.; Wahren, J.; Hendler, R.; Brundin, T. Splanchnic glucose and amino acid metabolism and obesity. J. Clin. Invest. 1974, 53, 582–590. [Google Scholar] [CrossRef]
- Gougeon, R.; Morais, J.A.; Chevalier, S.; Pereira, S.; Lamarche, M.; Marliss, E.B. Determinants of whole-body protein metabolism in subjects with and without type 2 diabetes. Diabetes Care 2008, 31, 128–133. [Google Scholar]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef]
- Cheng, S.; Rhee, E.P.; Larson, M.G.; Lewis, G.D.; McCabe, E.L.; Shen, D.; Palma, M.J.; Roberts, L.D.; Dejam, A.; Souza, A.L.; et al. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation 2012, 125, 2222–2231. [Google Scholar] [CrossRef]
- Ferrannini, E.; Natali, A.; Camastra, S.; Nannipieri, M.; Mari, A.; Adam, K.P.; Milburn, M.V.; Kastenmuller, G.; Adamski, J.; Tuomi, T.; et al. Early metabolic markers of the development of dysglycemia and type 2 diabetes and their physiological significance. Diabetes 2013, 62, 1730–1737. [Google Scholar] [CrossRef]
- Floegel, A.; Stefan, N.; Yu, Z.; Muhlenbruch, K.; Drogan, D.; Joost, H.G.; Fritsche, A.; Haring, H.U.; Hrabe de Angelis, M.; Peters, A.; et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 2013, 62, 639–648. [Google Scholar] [CrossRef]
- McCormack, S.E.; Shaham, O.; McCarthy, M.A.; Deik, A.A.; Wang, T.J.; Gerszten, R.E.; Clish, C.B.; Mootha, V.K.; Grinspoon, S.K.; Fleischman, A. Circulating branched-chain amino acid concentrations are associated with obesity and future insulin resistance in children and adolescents. Pediatr. Obes. 2013, 8, 52–61. [Google Scholar] [CrossRef]
- Wurtz, P.; Soininen, P.; Kangas, A.J.; Ronnemaa, T.; Lehtimaki, T.; Kahonen, M.; Viikari, J.S.; Raitakari, O.T.; Ala-Korpela, M. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 2013, 36, 648–655. [Google Scholar] [CrossRef]
- Menni, C.; Fauman, E.; Erte, I.; Perry, J.R.; Kastenmuller, G.; Shin, S.Y.; Petersen, A.K.; Hyde, C.; Psatha, M.; Ward, K.J.; et al. Biomarkers for type 2 diabetes and impaired fasting glucose using a non-targeted metabolomics approach. Diabetes 2013, in press. [Google Scholar]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O., Jr.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am. J. Med. 1980, 69, 491–497. [Google Scholar] [CrossRef]
- Eley, H.L.; Russell, S.T.; Tisdale, M.J. Effect of branched-chain amino acids on muscle atrophy in cancer cachexia. Biochem. J. 2007, 407, 113–120. [Google Scholar] [CrossRef]
- Beger, R.D. A review of applications of metabolomics in cancer. Metabolites 2013, 3, 552–574. [Google Scholar] [CrossRef]
- Milburn, M.V.; Lawton, K.A. Application of metabolomics to diagnosis of insulin resistance. Annu. Rev. Med. 2013, 64, 291–305. [Google Scholar] [CrossRef]
- Bhaskar, P.T.; Hay, N. The two TORCs and Akt. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef]
- Suzuki, T.; Inoki, K. Spatial regulation of the mTORC1 system in amino acids sensing pathway. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 671–679. [Google Scholar] [CrossRef]
- Wang, X.; Proud, C.G. Nutrient control of TORC1, a cell-cycle regulator. Trends Cell Biol. 2009, 19, 260–267. [Google Scholar] [CrossRef]
- Wang, X.; Proud, C.G. mTORC1 signaling: What we still don't know. J. Mol. Cell Biol. 2011, 3, 206–220. [Google Scholar] [CrossRef]
- Harper, A.E.; Miller, R.H.; Block, K.P. Branched-chain amino acid metabolism. Annu. Rev. Nutr. 1984, 4, 409–454. [Google Scholar] [CrossRef]
- Harris, R.A.; Zhang, B.; Goodwin, G.W.; Kuntz, M.J.; Shimomura, Y.; Rougraff, P.; Dexter, P.; Zhao, Y.; Gibson, R.; Crabb, D.W. Regulation of the branched-chain alpha-ketoacid dehydrogenase and elucidation of a molecular basis for maple syrup urine disease. Adv. Enzyme Regul. 1990, 30, 245–263. [Google Scholar] [CrossRef]
- Holecek, M. Relation between glutamine, branched-chain amino acids, and protein metabolism. Nutrition 2002, 18, 130–133. [Google Scholar] [CrossRef]
- Holecek, M. Branched-chain amino acid oxidation in skeletal muscle—physiology and clinical importance of its modulaton by reactant availability. Curr. Nutr. Food Sci. 2011, 7, 50–56. [Google Scholar] [CrossRef]
- Holecek, M.; Skopec, F.; Sprongl, L.; Mraz, J.; Skalska, H.; Pecka, M. Effect of alanyl-glutamine on leucine and protein metabolism in irradiated rats. Amino Acids 2002, 22, 95–108. [Google Scholar] [CrossRef]
- Odessey, R.; Goldberg, A.L. Oxidation of leucine by rat skeletal muscle. Am. J. Physiol. 1972, 223, 1376–1383. [Google Scholar]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Felig, P. The glucose-alanine cycle. Metabolism 1973, 22, 179–207. [Google Scholar] [CrossRef]
- Huffman, K.M.; Shah, S.H.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.; Slentz, C.A.; Tanner, C.J.; Kuchibhatla, M.; Houmard, J.A.; Newgard, C.B.; et al. Relationships between circulating metabolic intermediates and insulin action in overweight to obese, inactive men and women. Diabetes Care 2009, 32, 1678–1683. [Google Scholar] [CrossRef]
- Tai, E.S.; Tan, M.L.; Stevens, R.D.; Low, Y.L.; Muehlbauer, M.J.; Goh, D.L.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Lee, J.J.; et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia 2010, 53, 757–767. [Google Scholar] [CrossRef]
- Krebs, M.; Krssak, M.; Bernroider, E.; Anderwald, C.; Brehm, A.; Meyerspeer, M.; Nowotny, P.; Roth, E.; Waldhausl, W.; Roden, M. Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes 2002, 51, 599–605. [Google Scholar] [CrossRef]
- Kimball, S.R.; Jefferson, L.S. Signaling pathways and molecular mechanisms through which branched-chain amino acids mediate translational control of protein synthesis. J. Nutr. 2006, 136, 227S–231S. [Google Scholar]
- Dodd, K.M.; Tee, A.R. Leucine and mTORC1: A complex relationship. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1329–E1342. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; D’Alessio, D.; Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006, 3, 393–402. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Lewis, C.; Griffiths, B.; Schulze, A. A new player in the orchestra of cell growth: SREBP activity is regulated by mTORC1 and contributes to the regulation of cell and organ size. Biochem. Soc. Trans. 2009, 37, 278–283. [Google Scholar] [CrossRef]
- Long, W.; Saffer, L.; Wei, L.; Barrett, E.J. Amino acids regulate skeletal muscle PHAS-I and p70 S6-kinase phosphorylation independently of insulin. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E301–E306. [Google Scholar]
- Zick, Y. Ser/Thr phosphorylation of IRS proteins: A molecular basis for insulin resistance. Sci. STKE 2005, 2005, pe4. [Google Scholar]
- Melnik, B.C.; John, S.M.; Carrera-Bastos, P.; Cordain, L. The impact of cow’s milk-mediated mTORC1-signaling in the initiation and progression of prostate cancer. Nutr. Metab. (Lond.) 2012, 9. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef]
- Li, M.; Li, C.; Allen, A.; Stanley, C.A.; Smith, T.J. The structure and allosteric regulation of mammalian glutamate dehydrogenase. Arch. Biochem. Biophys. 2012, 519, 69–80. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Baracos, V.E.; Mackenzie, M.L. Investigations of branched-chain amino acids and their metabolites in animal models of cancer. J. Nutr. 2006, 136, 237S–242S. [Google Scholar]
- Warburg, O. Uber den Stoffwechsel der Carcinomzelle. Klinische Worchenschrift 1925, 4, 534–536. (in German). [Google Scholar] [CrossRef]
- De Blaauw, I.; Deutz, N.E.; von Meyenfeldt, M.F. Metabolic changes of cancer cachexia—second of two parts. Clin. Nutr. 1997, 16, 223–228. [Google Scholar] [CrossRef]
- Tisdale, M.J. Biology of cachexia. J. Natl. Cancer Inst. 1997, 89, 1763–1773. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Tennant, D.A.; Duran, R.V.; Boulahbel, H.; Gottlieb, E. Metabolic transformation in cancer. Carcinogenesis 2009, 30, 1269–1280. [Google Scholar] [CrossRef]
- Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 1955, 122, 501–514. [Google Scholar]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar]
- Mackenzie, M.L.; Baracos, V.E. Cancer-Associated Cachexia: Altered Metabolism of Protein and Amino Acids. In Metablic and Therapeutic Aspects of Amino Acids in Clinical Nutrition; Cynober, L.A., Ed.; CRC Press: Boca Raton, FL, USA, 2003; pp. 339–354. [Google Scholar]
- Lam, V.W.; Poon, R.T. Role of branched-chain amino acids in management of cirrhosis and hepatocellular carcinoma. Hepatol. Res. 2008, 38, S107–S115. [Google Scholar] [CrossRef]
- Beck, S.A. Nitrogen excretion in cancer cachexia and its modifications by a high fat diet in mice. Cancer Res. 1989, 49, 3800–3804. [Google Scholar]
- Norton, J.A.; Gorschlboth, C.M.; Wesely, R.A.; Burt, M.E.; Brennan, M.F. Fasting plasma amino acid levels in cancer patients. Cancer 1985, 56, 1181–1186. [Google Scholar] [CrossRef]
- Shiraki, M.; Shimomura, Y.; Miwa, Y.; Fukushima, H.; Murakami, T.; Tamura, T.; Tamura, N.; Moriwaki, H. Activation of hepatic branched-chain alpha-keto acid dehydrogenase complex by tumor necrosis factor-alpha in rats. Biochem. Biophys. Res. Commun. 2005, 328, 973–978. [Google Scholar] [CrossRef]
- Hunter, D.C.; Weintraub, M.; Blackburn, G.L.; Bistrian, B.R. Branched chain amino acids as the protein component of parenteral nutrition in cancer cachexia. Br. J. Surg. 1989, 76, 149–153. [Google Scholar] [CrossRef]
- Laviano, A.; Muscaritoli, M.; Cascino, A.; Preziosa, I.; Inui, A.; Mantovani, G.; Rossi-Fanelli, F. Branched-chain amino acids: The best compromise to achieve anabolism? Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 408–414. [Google Scholar] [CrossRef]
- Tayek, J.A.; Bistrian, B.R.; Hehir, D.J.; Martin, R.; Moldawer, L.L.; Blackburn, G.L. Improved protein kinetics and albumin synthesis by branched chain amino acid-enriched total parenteral nutrition in cancer cachexia. A prospective randomized crossover trial. Cancer 1986, 58, 147–157. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ikeda, K.; Arase, Y.; Suzuki, Y.; Suzuki, F.; Akuta, N.; Hosaka, T.; Murashima, N.; Saitoh, S.; Someya, T.; et al. Inhibitory effect of branched-chain amino acid granules on progression of compensated liver cirrhosis due to hepatitis C virus. J. Gastroenterol. 2008, 43, 63–70. [Google Scholar] [CrossRef]
- Muto, Y.; Sato, S.; Watanabe, A.; Moriwaki, H.; Suzuki, K.; Kato, A.; Kato, M.; Nakamura, T.; Higuchi, K.; Nishiguchi, S.; et al. Effects of oral branched-chain amino acid granules on event-free survival in patients with liver cirrhosis. Clin. Gastroenterol. Hepatol. 2005, 3, 705–713. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Asahini, Y.; Izumi, N. Long time oral supplementation with branched chain amino acids improves survivial and decreases recurrences in patients with hepatocellular carcinoma. Nihon Shokakibyo Zasshi 2008, 105, 808–816. [Google Scholar]
- Kawaguchi, T.; Nagao, Y.; Matsuoka, H.; Ide, T.; Sata, M. Branched-chain amino acid-enriched supplementation improves insulin resistance in patients with chronic liver disease. Int. J. Mol. Med. 2008, 22, 105–112. [Google Scholar]
- Nishitani, S.; Takehana, K.; Fujitani, S.; Sonaka, I. Branched-chain amino acids improve glucose metabolism in rats with liver cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1292–G1300. [Google Scholar] [CrossRef]
- Sato, S.; Watanabe, A.; Muto, Y.; Suzuki, K.; Kato, A.; Moriwaki, H.; Kato, M.; Nakamura, T. Clinical comparison of branched-chain amino acid (l-Leucine, l-Isoleucine, l-Valine) granules and oral nutrition for hepatic insufficiency in patients with decompensated liver cirrhosis (LIV-EN study). Hepatol Res. 2005, 31, 232–240. [Google Scholar] [CrossRef]
- Tomiya, T.; Inoue, Y.; Yanase, M.; Arai, M.; Ikeda, H.; Tejima, K.; Nagashima, K.; Nishikawa, T.; Fujiwara, K. Leucine stimulates the secretion of hepatocyte growth factor by hepatic stellate cells. Biochem. Biophys. Res. Commun. 2002, 297, 1108–1111. [Google Scholar] [CrossRef]
- Tsubouchi, H. Hepatocyte growth factor for liver disease. Hepatology 1999, 30, 333–334. [Google Scholar] [CrossRef]
- Miuma, S.; Ichikawa, T.; Arima, K.; Takeshita, S.; Muraoka, T.; Matsuzaki, T.; Ootani, M.; Shibata, H.; Akiyama, M.; Ozawa, E.; et al. Branched-chain amino acid deficiency stabilizes insulin-induced vascular endothelial growth factor mRNA in hepatocellular carcinoma cells. J. Cell Biochem. 2012, 113, 3113–3121. [Google Scholar] [CrossRef]
- Hagiwara, A.; Nishiyama, M.; Ishizaki, S. Branched-chain amino acids prevent insulin-induced hepatic tumor cell proliferation by inducing apoptosis through mTORC1 and mTORC2-dependent mechanisms. J. Cell Physiol. 2012, 227, 2097–2105. [Google Scholar] [CrossRef]
- Tom, A.; Nair, K.S. Assessment of branched-chain amino acid status and potential for biomarkers. J. Nutr. 2006, 136, 324S–330S. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
O'Connell, T.M. The Complex Role of Branched Chain Amino Acids in Diabetes and Cancer. Metabolites 2013, 3, 931-945. https://doi.org/10.3390/metabo3040931
O'Connell TM. The Complex Role of Branched Chain Amino Acids in Diabetes and Cancer. Metabolites. 2013; 3(4):931-945. https://doi.org/10.3390/metabo3040931
Chicago/Turabian StyleO'Connell, Thomas M. 2013. "The Complex Role of Branched Chain Amino Acids in Diabetes and Cancer" Metabolites 3, no. 4: 931-945. https://doi.org/10.3390/metabo3040931
APA StyleO'Connell, T. M. (2013). The Complex Role of Branched Chain Amino Acids in Diabetes and Cancer. Metabolites, 3(4), 931-945. https://doi.org/10.3390/metabo3040931