
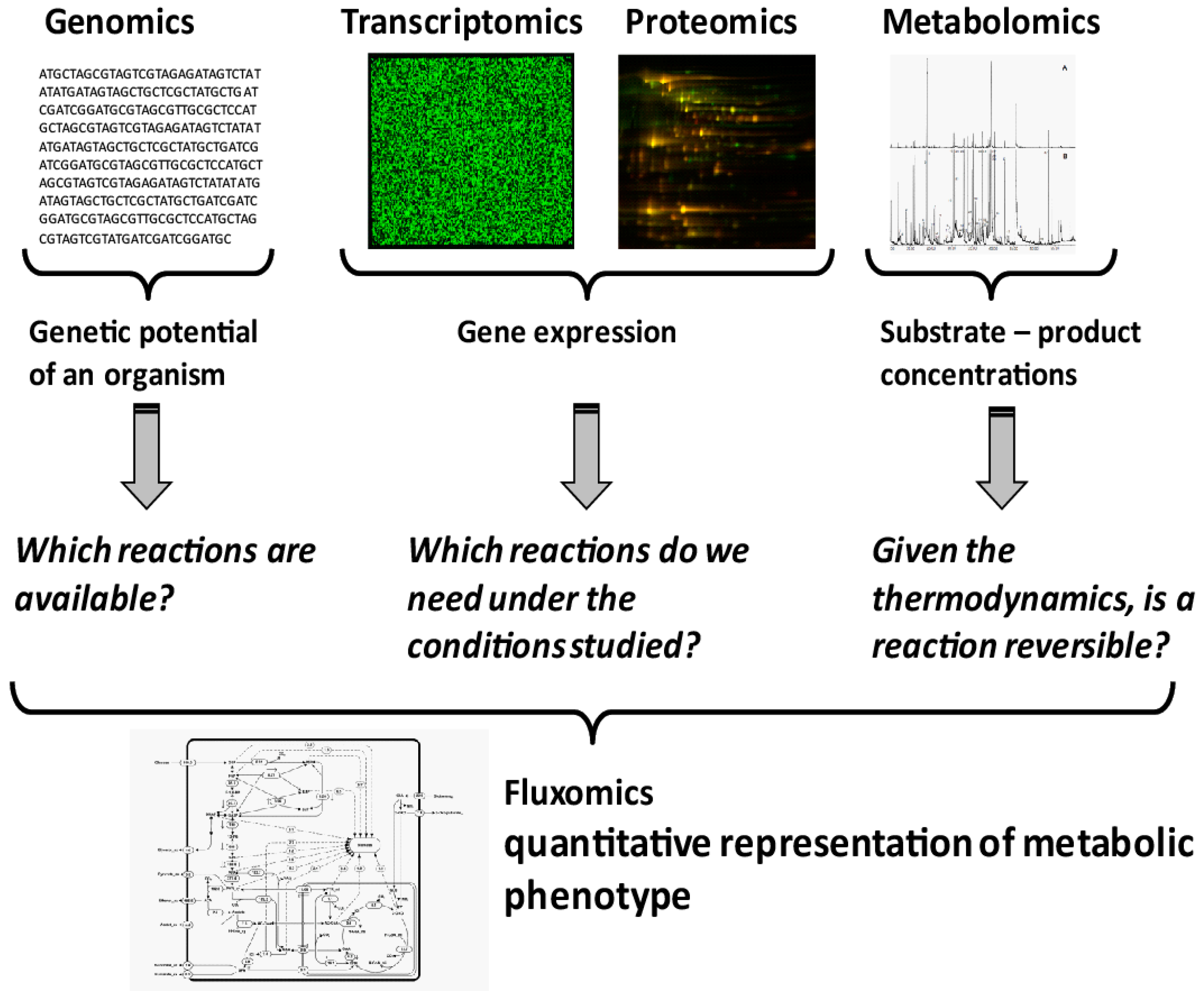
Quantification of Microbial Phenotypes


Abstract
:1. Introduction
2. Quantitative Analysis of Microbial Metabolites
2.1. High-Performance Liquid Chromatography (HPLC)
2.2. Gas Chromatography-Mass Spectrometry (GC-MS)
2.3. Liquid Chromatography Electro-Spray Ionization Tandem Mass Spectrometry (LC-ESI-MS/MS)
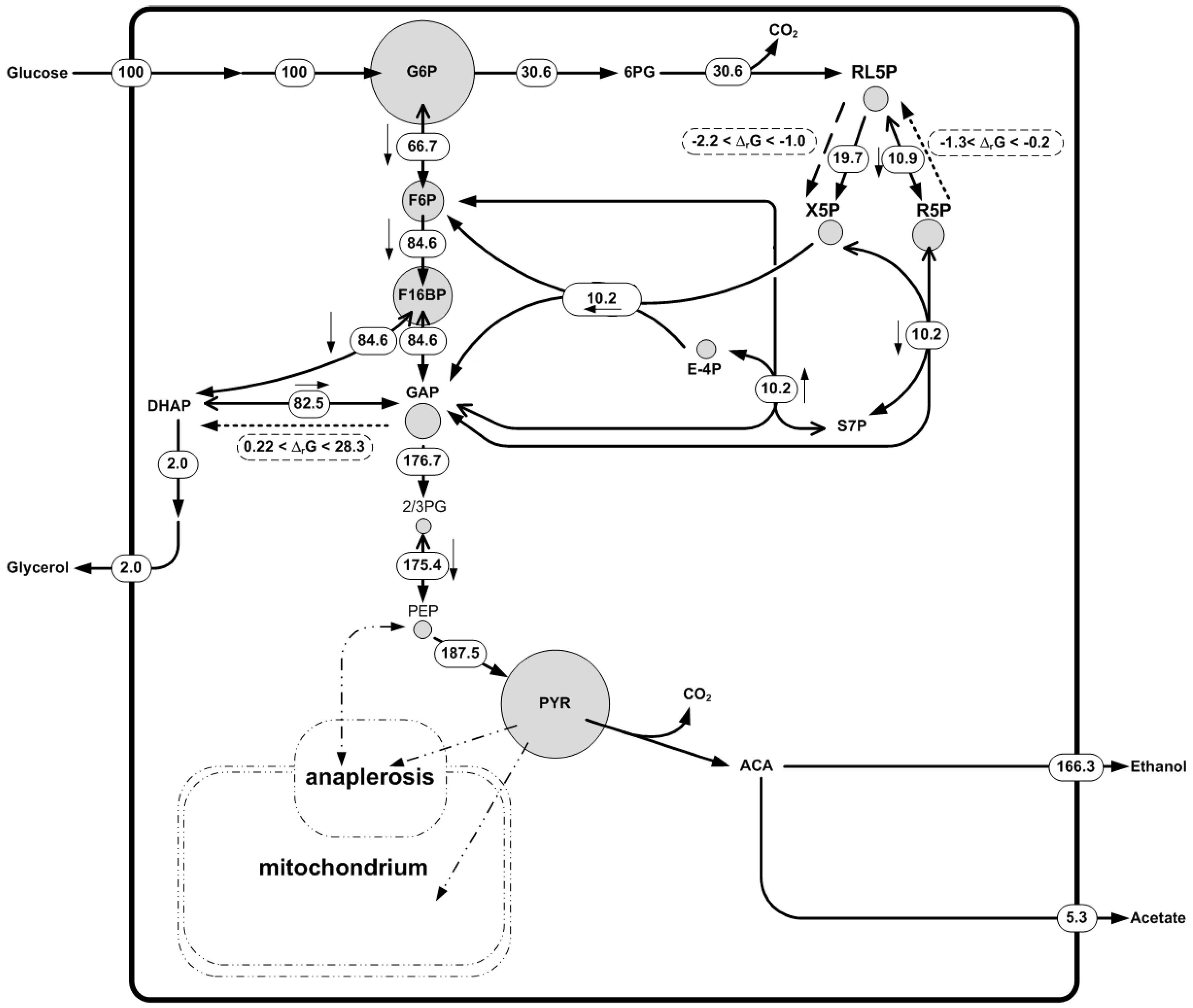
3. Thermodynamics and Metabolomics Integration into Metabolic Networks
3.1. Second Law of Thermodynamics and Reactions Directionality
- The first law is the conservation law which states that energy can neither be created nor destroyed. In a closed system energy is constant.
- The second law states that spontaneous natural processes increase the overall entropy of the universe.
3.2. ΔfG0 for Physiological Conditions
3.3. ΔfG0 for Reactants as Groups of Species
3.4. Gibbs Energy of Transport Reactions
3.5. Example 1: Standard Transformed Gibbs Energies of Yeast Glycolysis Reactions at Physiological Conditions
3.6. Application of Network Thermodynamics to Large-Scale Models
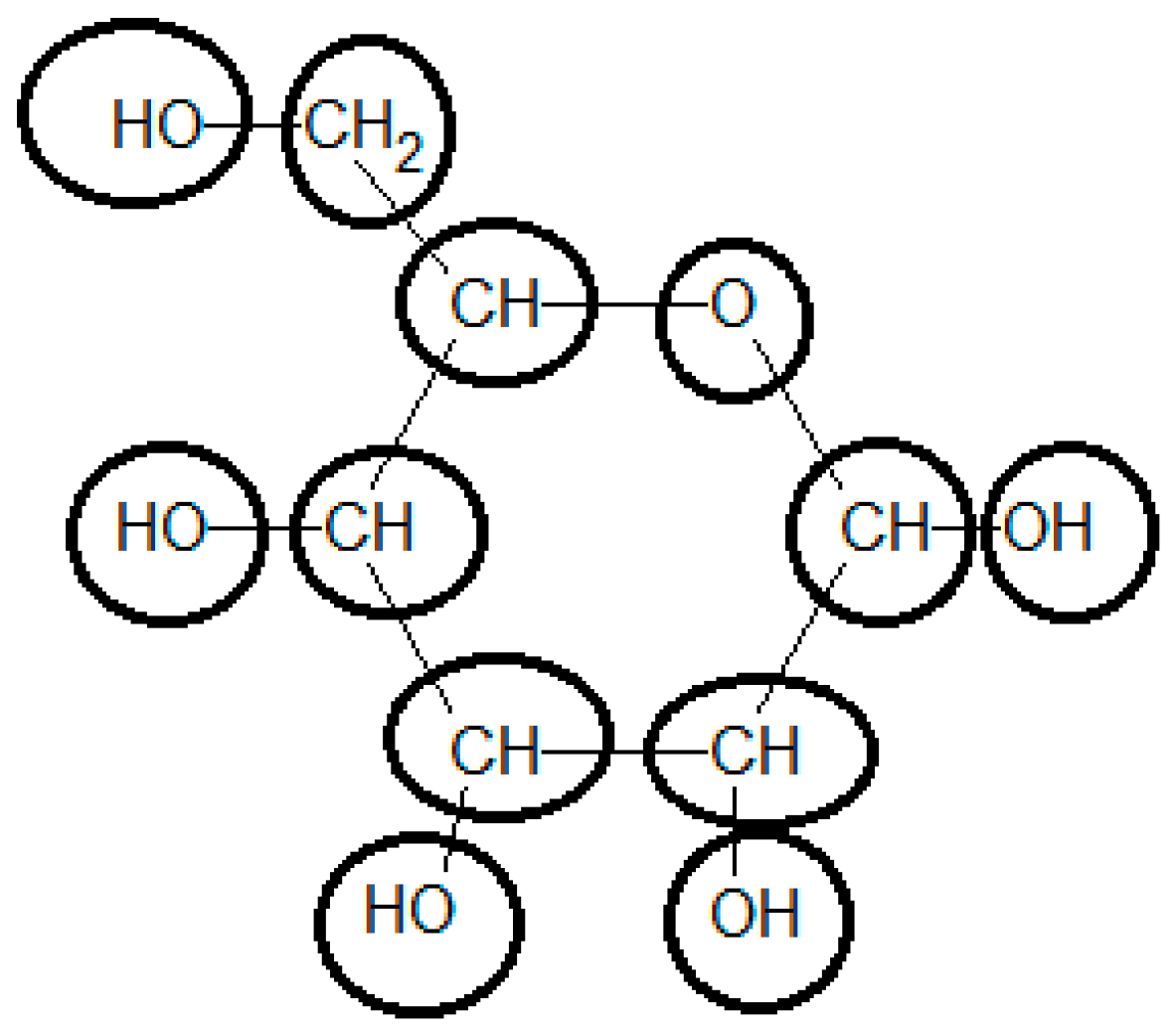
3.7. Example 2: Standard Gibbs Energy of Formation of Glucose Estimated by the Group Contribution Method
3.8. Incorporation of Quantitative Metabolite Data into Metabolic Networks
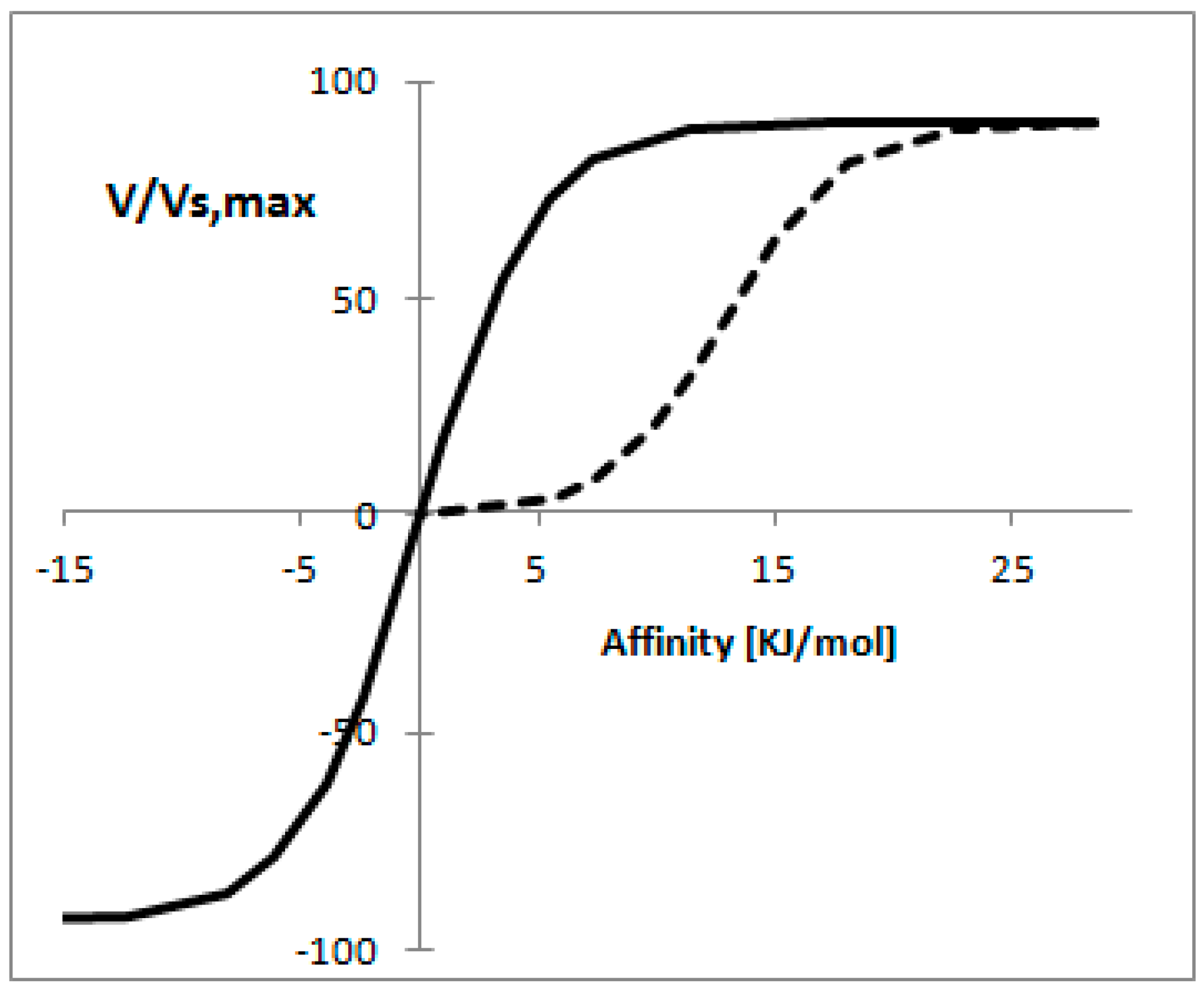
3.9. Non-Equilibrium Thermodynamics for Enzymatic Reactions
4. Quantification of Metabolic Phenotype Using 13C Fluxomics
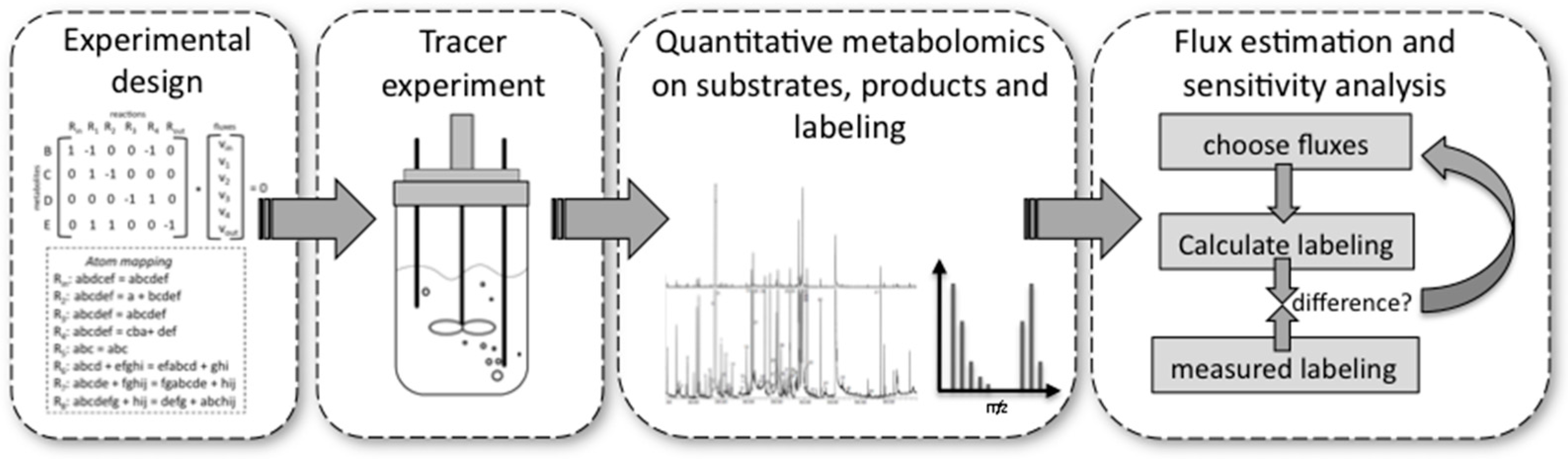
4.1. Performing a 13C Metabolic Flux Analysis
- Experimental design is important because very costly tracer substrates can be ineffective when used in a suboptimal design. For design, the stoichiometry and the atom transitions must be known. By performing labelling experiments in silico for the expected range of fluxes, it is possible to determine the most suitable labelled substrate(s) to use and the most suitable labelled metabolites to analyse by MS or NMR, e.g., the metabolites for which labelling patterns are most responsive to changes in fluxes. These metabolites can be derived from macromolecules, such as proteinogenic amino acids or free intracellular metabolites. The choice depends on available sample size and equipment sensitivities.
- Performing the experiment: Pseudo-steady state conditions require balanced growth throughout the tracer experiment. It is essential that no nutrient limitations, for example oxygen in aerobic experiments, occur during the experiment. Biomass composition should remain fairly constant and the mass balances of the system for at least one macro-nutrient, for example carbon or nitrogen, should be closed. Bioreactors guarantee such a controlled environment with balanced growth, but it has been shown that shake flasks and microplate systems can also deliver these conditions within certain limits [76].
- Quantitative metabolomics: All major substrates and products need to be accurately quantified. For MFA the biomass itself is an important product. Its major composition (DNA/RNA, protein, carbohydrate, lipid contents) determines a whole range of anabolic fluxes. Finally, MS [77] or NMR [70] equipment are required to quantify the labelling enrichment in the target metabolites (e.g., GC/MS analysis of proteinogenic amino acids). Accurate isotope ratios, although not absolute quantification, of those metabolites is essential.
- Flux estimation and sensitivity analysis: Flux estimation is an iterative process in which the stoichiometric and atom mapping networks are used to calculate labelling outputs while achieving the experimentally observed uptake, secretion, and growth rates. At each iteration the calculated labelling outputs are compared to the experimentally determined ones and the resulting fluxes are updated until the differences between calculated and measured labellings are minimized. At the end of this fitting process, the sensitivity of fluxes is further evaluated using statistical procedures. As a result, each flux is obtained with a certain confidence interval.
4.2. Example 3: Integration of Quantitative Metabolite Data, Thermodynamics and 13C Fluxomics Based on Saccharomyces Cerevisiae GeM
- reactions catalyzed by different enzymes in either direction should be lumped together resulting in a reversible reaction, otherwise a thermodynamic equilibrium state with a zero Gibbs energy of reaction will be considered for both of them;
- in thermodynamics, reactions are described in terms of reactants instead of species. For example the species HCO3−, CO32−, CO2, and H2CO3 are grouped as the reactant CO2tot because aqueous phase carbon dioxide is distributed in those species;
- for reactions that contain CO2tot, H2O was added to the opposite side of the reaction in order to balance oxygen atoms; and
- the oxidized and reduced flavin adenine dinucleotide (FAD) in mitochondria were replaced by oxidized and reduced FADenz in order to represent the enzyme-bound FAD cofactor.
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, A.; Hurlebaus, J.; Wandrey, C.; Takors, R. Metabolomics: Quantification of intracellular metabolite dynamics. Biomol. Eng. 2002, 19, 5–15. [Google Scholar] [CrossRef]
- De Koning, W.; van Dam, K. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 1992, 204, 118–123. [Google Scholar] [CrossRef]
- Theobald, U.; Mailinger, W.; Baltes, M.; Rizzi, M.; Reuss, M. In vivo analysis of metabolic dynamics in saccharomyces cerevisiae: I. Experimental observations. Biotechnol. Bioeng. 1997, 55, 305–316. [Google Scholar] [CrossRef]
- Bolten, C.J.; Wittmann, C. Appropriate sampling for intracellular amino acid analysis in five phylogenetically different yeasts. Biotechnol. Lett. 2008, 30, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.; Krömer, J.O.; Kiefer, P.; Binz, T.; Heinzle, E. Impact of the cold shock phenomenon on quantification of intracellular metabolites in bacteria. Anal. Biochem. 2004, 327, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—the link between genotypes and phenotypes. Plant. Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, D.Y.; Kim, T.Y. Systems biotechnology for strain improvement. Trends Biotechnol. 2005, 23, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Krömer, J.O.; Fritz, M.; Heinzle, E.; Wittmann, C. In vivo quantification of intracellular amino acids and intermediates of the methionine pathway in Corynebacterium glutamicum. Anal. Biochem. 2005, 340, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Dietmair, S.; Timmins, N.E.; Gray, P.P.; Nielsen, L.K.; Krömer, J.O. Towards quantitative metabolomics of mammalian cells: Development of a metabolite extraction protocol. Anal. Biochem. 2010, 404, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, E.; Nielsen, L.K.; Abeydeera, P.; Krömer, J.O. Quantitative analysis of intracellular sugar phosphates and sugar nucleotides in encapsulated streptococci using hpaec-pad. Biotechnol. J. 2009, 4, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kanani, H.H.; Klapa, M.I. Data correction strategy for metabolomics analysis using gas chromatography-mass spectrometry. Metab. Eng. 2007, 9, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Anal. Chem. 2007, 79, 7554–7559. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Groenke, K.; Takors, R.; Wandrey, C.; Oldiges, M. Simultaneous determination of multiple intracellular metabolites in glycolysis, pentose phosphate pathway and tricarboxylic acid cycle by liquid chromatography-mass spectrometry. J. Chromatogr. A 2007, 1147, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Matrix effect in quantitative LC/MS/MS analyses of biological fluids: A method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 1998, 70, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Mashego, M.R.; Wu, L.; van Dam, J.C.; Ras, C.; Vinke, J.L.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Miracle: Mass isotopomer ratio analysis of U-13C-labeled extracts. A new method for accurate quantification of changes in concentrations of intracellular metabolites. Biotechnol. Bioeng. 2004, 85, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; van Dien, S.J.; Rabinowitz, J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Kummel, A.; Panke, S.; Heinemann, M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamics-based metabolic flux analysis. Biophys. J. 2007, 92, 1792–1805. [Google Scholar] [CrossRef] [PubMed]
- Matuszczyk, J.C.; Teleki, A.; Pfizenmaier, J.; Takors, R. Compartment-specific metabolomics for Cho reveals that ATP pools in mitochondria are much lower than in cytosol. Biotechnol. J. 2015, 10, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.M.T.; Thiele, I.; Nasheuer, H.P. Quantitative assignment of reaction directionality in constraint-based models of metabolism: Application to Escherichia coli. Biophys. Chem. 2009, 145, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Martínez, V.S.; Quek, L.E.; Nielsen, L.K. Network thermodynamic curation of human and yeast genome-scale metabolic models. Biophys. J. 2014, 107, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Yang, L.; Mahadevan, R. Thermodynamic analysis of regulation in metabolic networks using constraint-based modeling. BMC Res. Notes 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdottir, H.S.; Thiele, I.; Fleming, R.M.T. Quantitative assignment of reaction directionality in a multicompartmental human metabolic reconstruction. Biophys. J. 2012, 102, 1703–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberty, R.A. Thermodynamics of Biochemical Reactions; Wiley: New York, NY, USA, 2003; p. 397. [Google Scholar]
- Maskow, T.; von Stockar, U. How reliable are thermodynamic feasibility statements of biochemical pathways? Biotechnol. Bioeng. 2005, 92, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Vojinovic, V.; von Stockar, U. Influence of uncertainties in pH, PMG, activity coefficients, metabolite concentrations, and other factors on the analysis of the thermodynamic feasibility of metabolic pathways. Biotechnol. Bioeng. 2009, 103, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Alberty, R.A. Legendre transforms in chemical thermodynamics. Chem. Rev. 1994, 94, 1457–1482. [Google Scholar] [CrossRef]
- Henry, C.S.; Jankowski, M.D.; Broadbelt, L.J.; Hatzimanikatis, V. Genome-scale thermodynamic analysis of Escherichia coli metabolism. Biophys. J. 2006, 90, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Jol, S.J.; Kummel, A.; Hatzimanikatis, V.; Beard, D.A.; Heinemann, M. Thermodynamic calculations for biochemical transport and reaction processes in metabolic networks. Biophys. J. 2010, 99, 3139–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soh, K.C.; Hatzimanikatis, V. Network thermodynamics in the post-genomic Era. Curr. Opin. Microbiol. 2010, 13, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Mavrovouniotis, M.L. Group contributions for estimating standard gibbs energies of formation of biochemical-compounds in aqueous-solution. Biotechnol. Bioeng. 1990, 36, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Mavrovouniotis, M.L. Estimation of standard gibbs energy changes of biotransformations. J. Biol. Chem. 1991, 266, 14440–14445. [Google Scholar] [PubMed]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.O. A genome-scale metabolic reconstruction for Escherichia coli k-12 mg1655 that accounts for 1260 Orfs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, M.D.; Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 2008, 95, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Noor, E.; Bar-Even, A.; Flamholz, A.; Lubling, Y.; Davidi, D.; Milo, R. An integrated open framework for thermodynamics of reactions that combines accuracy and coverage. Bioinformatics 2012, 28, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Noor, E.; Haraldsdottir, H.S.; Milo, R.; Fleming, R.M.T. Consistent estimation of gibbs energy using component contributions. PLoS Comput. Biol. 2013, 9, e1003098. [Google Scholar] [CrossRef] [PubMed]
- Mavrovouniotis, M.L. Identification of localized and distributed bottlenecks in metabolic pathways. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1993, 1, 275–283. [Google Scholar] [PubMed]
- Finley, S.D.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamic analysis of biodegradation pathways. Biotechnol. Bioeng. 2009, 103, 532–541. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, D.; Gangoiti, J.A.; King, Z.A.; Naviaux, R.K.; Barshop, B.A.; Palsson, B.O.; Feist, A.M. A model-driven quantitative metabolomics analysis of aerobic and anaerobic metabolism in E. coli k-12 mg1655 that is biochemically and thermodynamically consistent. Biotechnol. Bioeng. 2014, 111, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.L.; Vo, T.D.; Schilling, C.H.; Palsson, B.O. An expanded genome-scale model of Escherichia coli k-12 (iJR904 GSM/GPR). Genome Biol. 2003, 4, R54. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, N.; Kummel, A.; Heinemann, M. Annet: A tool for network-embedded thermodynamic analysis of quantitative metabolome data. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, V.S.; Nielsen, L.K. Next: Integration of thermodynamic constraints and metabolomics data into a metabolic network. In Metabolic Flux Analysis; Springer: Berlin, Germany, 2014; pp. 65–78. [Google Scholar]
- Hoppe, A.; Hoffmann, S.; Holzhutter, H.G. Including metabolite concentrations into flux balance analysis: Thermodynamic realizability as a constraint on flux distributions in metabolic networks. BMC Syst. Biol. 2007, 1, 23. [Google Scholar] [CrossRef] [PubMed]
- Ataman, M.; Hatzimanikatis, V. Heading in the right direction: Thermodynamics-based network analysis and pathway engineering. Curr. Opin. Biotechnol. 2015, 36, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Boghigian, B.A.; Shi, H.; Lee, K.; Pfeifer, B.A. Utilizing elementary mode analysis, pathway thermodynamics, and a genetic algorithm for metabolic flux determination and optimal metabolic network design. BMC Syst. Biol. 2010, 4, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jol, S.J.; Kummel, A.; Terzer, M.; Stelling, J.; Heinemann, M. System-level insights into yeast metabolism by thermodynamic analysis of elementary flux modes. PLoS Comput. Biol. 2012, 8, e1002415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstl, M.P.; Jungreuthmayer, C.; Zanghellini, J. Tefma: Computing thermodynamically feasible elementary flux modes in metabolic networks. Bioinformatics 2015, 31, 2232–2234. [Google Scholar] [CrossRef] [PubMed]
- Gerstl, M.P.; Ruckerbauer, D.E.; Mattanovich, D.; Jungreuthmayer, C.; Zanghellini, J. Metabolomics integrated elementary flux mode analysis in large metabolic networks. Sci. Rep. 2015, 5, 8930. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, H.V.; Dam, K.V. Thermodynamics and Control of Biological Free-Energy Transduction; Elsevier: Amsterdam, The Netherland; Oxford, UK, 1987; p. 568. [Google Scholar]
- Rottenberg, H. The thermodynamic description of enzyme-catalyzed reactions: The linear relation between the reaction rate and the affinity. Biophys. J. 1973, 13, 503–511. [Google Scholar] [CrossRef]
- Vandermeer, R.; Westerhoff, H.V.; Vandam, K. Linear relation between rate and thermodynamic force in enzyme-catalyzed reactions. Biochim. Biophys. Acta 1980, 591, 488–493. [Google Scholar] [CrossRef]
- Nielsen, J. Metabolic control analysis of biochemical pathways based on a thermokinetic description of reaction rates. Biochem. J. 1997, 321, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Visser, D.; Heijnen, J.J. Dynamic simulation and metabolic re-design of a branched pathway using linlog kinetics. Metab. Eng. 2003, 5, 164–176. [Google Scholar] [CrossRef]
- Yu, X.; White, L.T.; Doumen, C.; Damico, L.A.; LaNoue, K.F.; Alpert, N.M.; Lewandowski, E.D. Kinetic analysis of dynamic 13C NMR spectra: Metabolic flux, regulation, and compartmentation in hearts. Biophys. J. 1995, 69, 2090–2102. [Google Scholar] [CrossRef]
- Vallino, J.J.; Stephanopoulos, G. Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol. Bioeng. 2000, 67, 872–885. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia-coli w3110. Appl. Environ. Microbiol. 1994, 60, 3724–3731. [Google Scholar]
- Leighty, R.W.; Antoniewicz, M.R. Dynamic metabolic flux analysis (DMFA): A framework for determining fluxes at metabolic non-steady state. Metab. Eng. 2011, 13, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Martínez, V.S.; Buchsteiner, M.; Gray, P.; Nielsen, L.K.; Quek, L.-E. Dynamic metabolic flux analysis using B-splines to study the effects of temperature shift on cho cell metabolism. Metab. Eng. Commun. 2015, 2, 46–57. [Google Scholar] [CrossRef]
- Marx, A.; deGraaf, A.A.; Wiechert, W.; Eggeling, L.; Sahm, H. Determination of the fluxes in the central metabolism of Corynebacterium glutamicum by nuclear magnetic resonance spectroscopy combined with metabolite balancing. Biotechnol. Bioeng. 1996, 49, 111–129. [Google Scholar] [CrossRef]
- Sonntag, K.; Eggeling, L.; Degraaf, A.A.; Sahm, H. Flux partitioning in the split pathway of lysine synthesis in Corynebacterium-glutamicum quantification by 13C-NMR and H-1-NMR spectroscopy. Eur. J. Biochem. 1993, 213, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W.; de Graaf, A.A. Bidirectional reaction steps in metabolic networks: I. Modeling and simulation of carbon isotope labeling experiments. Biotechnol. Bioeng. 1997, 55, 101–117. [Google Scholar] [CrossRef]
- Wiechert, W.; Siefke, C.; deGraaf, A.A.; Marx, A. Bidirectional reaction steps in metabolic networks: II. Flux estimation and statistical analysis. Biotechnol. Bioeng. 1997, 55, 118–135. [Google Scholar] [CrossRef]
- Iwatani, S.; Yamada, Y.; Usuda, Y. Metabolic flux analysis in biotechnology processes. Biotechnol. Lett. 2008, 30, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.J.; Martin, H.G.; Myers, S.; Rodriguez, S.; Baidoo, E.E.K.; Keasling, J.D. Advances in analysis of microbial metabolic fluxes via 13C isotopic labeling. Mass Spectr. Rev. 2009, 28, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W. 13C metabolic flux analysis. Metab. Eng. 2001, 3, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W.; Mollney, M.; Petersen, S.; de Graaf, A.A. A universal framework for 13C metabolic flux analysis. Metab. Eng. 2001, 3, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Sauer, U.; Hatzimanikatis, V.; Bailey, J.E.; Hochuli, M.; Szyperski, T.; Wuthrich, K. Metabolic fluxes in riboflavin-producing bacillus subtilis. Nat. Biotechnol. 1997, 15, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Gombert, A.K.; dos Santos, M.M.; Christensen, B.; Nielsen, J. Network identification and flux quantification in the central metabolism of saccharomyces cerevisiae under different conditions of glucose repression. J. Bacteriol. 2001, 183, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Krömer, J.O.; Bolten, C.J.; Heinzle, E.; Schroder, H.; Wittmann, C. Physiological response of Corynebacterium glutamicum to oxidative stress induced by deletion of the transcriptional repressor mcbr. Microbiol. SGM 2008, 154, 3917–3930. [Google Scholar] [CrossRef] [PubMed]
- Krömer, J.O.; Sorgenfrei, O.; Klopprogge, K.; Heinzle, E.; Wittmann, C. In-depth profiling of lysine-producing Corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J. Bacteriol. 2004, 186, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Millard, P.; Portais, J.C.; Mendes, P. Impact of kinetic isotope effects in isotopic studies of metabolic systems. BMC Syst. Biol. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, T.E.; Long, C.P.; Gonzalez, J.E.; Feist, A.M.; Antoniewicz, M.R.; Palsson, B.O. Evolution of E. coli on [U-13C] glucose reveals a negligible isotopic influence on metabolism and physiology. PLoS ONE 2016, 11, e0151130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittmann, C.; Kim, H.M.; Heinzle, E. Metabolic network analysis of lysine producing Corynebacterium glutamicum at a miniaturized scale. Biotechnol. Bioeng. 2004, 87, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.; Nielsen, J. Isotopomer analysis using GC-MS. Metab. Eng. 1999, 1, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Elementary metabolite units (EMU): A novel framework for modeling isotopic distributions. Metab. Eng. 2007, 9, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.E.; Wittmann, C.; Nielsen, L.K.; Krömer, J.O. Openflux: Efficient modelling software for 13C-based metabolic flux analysis. Microb. Cell Fact. 2009, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, N.; Fischer, E.; Sauer, U. Fiatflux—A software for metabolic flux analysis from 13C-glucose experiments. BMC Bioinform. 2005, 6, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.; Antoniewicz, M.R.; Stephanopoulos, G.; Kelleher, J.K. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J. Biol. Chem. 2008, 283, 20621–20627. [Google Scholar] [CrossRef] [PubMed]
- Millard, P.; Sokol, S.; Letisse, F.; Portais, J.C. Isodesign: A software for optimizing the design of 13C-metabolic flux analysis experiments. Biotechnol. Bioeng. 2014, 111, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, M.; Noh, K.; Dalman, T.; Niedenfuhr, S.; Stute, B.; Wiechert, W. 13CFLUX2-high-performance software suite for 13C-metabolic flux analysis. Bioinformatics 2013, 29, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D. Inca: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Kajihata, S.; Furusawa, C.; Matsuda, F.; Shimizu, H. Openmebius: An open source software for isotopically nonstationary 13C-based metabolic flux analysis. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Canelas, A.B.; van Gulik, W.M.; Heijnen, J.J. Determination of the cytosolic free nad/nadh ratio in saccharomyces cerevisiae under steady-state and highly dynamic conditions. Biotechnol. Bioeng. 2008, 100, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.; Hans, M.; Heinzle, E. In vivo analysis of intracellular amino acid labelings by GC/MS. Anal. Biochem. 2002, 307, 379–382. [Google Scholar] [CrossRef]
- Heavner, B.D.; Smallbone, K.; Barker, B.; Mendes, P.; Walker, L.P. Yeast 5-an expanded reconstruction of the saccharomyces cerevisiae metabolic network. BMC Syst. Biol. 2012, 6, 55. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | ΔfG0 [kJ/mol] | Charge (zi) | Number of H Atoms (NH) | ΔfG′0 [kJ/mol] |
|---|---|---|---|---|
| 3-Phospho-glyceroyl phosphate | −2356.14 | −4 | 4 | −2206.35 |
| −2401.58 | −3 | 5 | ||
| 2-Phospho-glycerate | −1496.38 | −3 | 4 | −1341.51 |
| −1539.99 | −2 | 5 | ||
| 3-Phospho-glycerate | −1502.54 | −3 | 4 | −1347.41 |
| −1545.52 | −2 | 5 | ||
| ADP | −1906.13 | −3 | 12 | −1425.17 |
| −1947.1 | −2 | 13 | ||
| −1971.98 | −1 | 14 | ||
| ATP | −2768.1 | −4 | 12 | −2292.28 |
| −2811.48 | −3 | 13 | ||
| −2838.18 | −2 | 14 | ||
| Glycerone phosphate | −1296.26 | −2 | 5 | −1095.82 |
| −1328.8 | −1 | 6 | ||
| Fructose 6-phosphate | −1760.8 | −2 | 11 | −1316.55 |
| −1796.6 | −1 | 12 | ||
| Fructose 1,6-bisphosphate | −2601.4 | −4 | 10 | −2206.14 |
| −2639.36 | −3 | 11 | ||
| −2673.89 | −2 | 12 | ||
| Glyceraldehyde 3-phosphate | −1288.6 | −2 | 5 | −1088.16 |
| −1321.14 | −1 | 6 | ||
| Glucose | −915.9 | 0 | 12 | −428.06 |
| Glucose 6-phosphate | −1763.94 | −2 | 11 | −1319.75 |
| −1800.59 | −1 | 12 | ||
| H2O | −237.19 | 0 | 2 | −155.88 |
| NAD | 0 | −1 | 26 | 1056.29 |
| NADH | 22.65 | −2 | 27 | 1117.50 |
| Phosphoenolpyruvate | −1263.65 | −3 | 2 | −1189.04 |
| −1303.61 | −2 | 3 | ||
| Pi | −1096.1 | −2 | 1 | −1059.30 |
| −1137.3 | −1 | 2 | ||
| Pyruvate | −472.27 | −1 | 3 | −351.01 |
| rxnID | Extended Reaction | ΔrG′0 (kJ/mol) |
|---|---|---|
| HEX1 | Glucose + ATP = Glucose 6-phosphate + ADP | −24.58 |
| PGI | Glucose 6-phosphate = Fructose 6-phosphate | 3.20 |
| PFK | Fructose 6-phosphate + ATP = Fructose 1,6-bisphosphate + ADP | −22.48 |
| FBA | Fructose 1,6-bisphosphate = Glycerone phosphate + Glyceraldehyde 3-phosphate | 22.15 |
| TPI | Glycerone phosphate = Glyceraldehyde 3-phosphate | 7.66 |
| GAPD | Glyceraldehyde 3-phosphate + Pi + NAD = NADH + 3-Phospho-glyceroyl phosphate | 2.32 |
| PGK | 3-Phospho-glyceroyl phosphate + ADP = 3-Phospho-glycerate + ATP | 8.16 |
| PGM | 3-Phospho-glycerate = 2-Phospho-glycerate | 5.90 |
| ENO | 2-Phospho-glycerate = Phosphoenolpyruvate + H2O | −3.41 |
| PYK | Phosphoenolpyruvate + ADP = Pyruvate + ATP | −29.08 |
| Group | Contribution 1 [kJ/mol] | Contribution 2 [kJ/mol] | # Occurrences | Total Contribution 1 [kJ/mol] | Total Contribution 2 [kJ/mol] |
|---|---|---|---|---|---|
| Ori | −103.4 | 0.0 | 1 | −103.4 | 0.0 |
| OH- (secondary) | −131.5 | −173.8 | 4 | −525.9 | −695.0 |
| -O- (ring) | −101.7 | −153.2 | 1 | −101.7 | −153.2 |
| >CH2 | 7.1 | 6.8 | 1 | 7.1 | 6.8 |
| >CH- ( ring) | −10.9 | 20.3 | 5 | −54.4 | 101.3 |
| OH- (primary) | −119.7 | −173.8 | 1 | −119.7 | −173.8 |
| Total | - | - | - | −898.07 | −913.89 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, V.S.; Krömer, J.O. Quantification of Microbial Phenotypes. Metabolites 2016, 6, 45. https://doi.org/10.3390/metabo6040045
Martínez VS, Krömer JO. Quantification of Microbial Phenotypes. Metabolites. 2016; 6(4):45. https://doi.org/10.3390/metabo6040045
Chicago/Turabian StyleMartínez, Verónica S., and Jens O. Krömer. 2016. "Quantification of Microbial Phenotypes" Metabolites 6, no. 4: 45. https://doi.org/10.3390/metabo6040045
APA StyleMartínez, V. S., & Krömer, J. O. (2016). Quantification of Microbial Phenotypes. Metabolites, 6(4), 45. https://doi.org/10.3390/metabo6040045