
Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease

 , ,
, ,  and
and 
Abstract
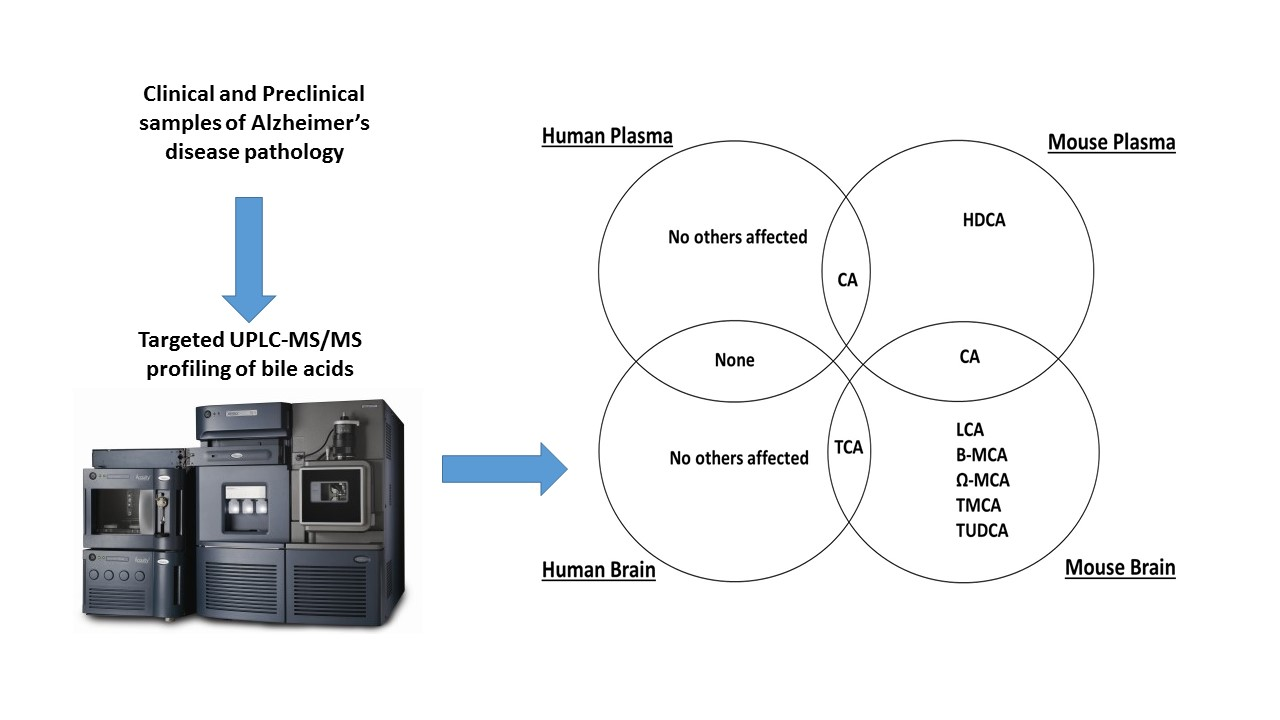
:
1. Introduction
2. Results
2.1. Bile Acid Levels in Human Plasma
2.2. Bile Acid Levels in Mouse Plasma
2.3. Bile Acid Levels in Human Brain
2.4. Bile Acid Levels in Mouse Brain
3. Discussion
4. Materials and Methods
4.1. Human Plasma and Post-Mortem Brain Tissue
4.2. Mouse Plasma and Brain Tissue
4.3. Brain Tissue Extraction
4.4. Bile Acid Quantifications
5. Statistical Analysis
6. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Schoonjans, K. TGR5 and immunometabolism: Insights from physiology and pharmacology. Trends Pharmacol. Sci. 2015, 36, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Donkin, J.; Wellington, C. Greasing the wheels of Abeta clearance in Alzheimer’s disease: The role of lipids and apolipoprotein E. Biofactors 2009, 35, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Ogundare, M.; Theofilopoulos, S.; Lockhart, A.; Hall, L.J.; Arenas, E.; Sjovall, J.; Brenton, A.G.; Wang, Y.; Griffiths, W.J. Cerebrospinal fluid steroidomics: Are bioactive bile acids present in brain? J. Biol. Chem. 2010, 285, 4666–4679. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, N.; Grassano, A.; Thambisetty, M.; Lovestone, S.; Legido-Quigley, C. A proposed metabolic strategy for monitoring disease progression in Alzheimer’s disease. Electrophoresis 2009, 30, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Olazarán, J.; Gil-de-Gómez, L.; Rodríguez-Martín, A.; Valentí-Soler, M.; Frades-Payo, B.; Marín-Muñoz, J.; Antúnez, C.; Frank-García, A.; Acedo-Jiménez, C.; Morlán-Gracia, L.; et al. A blood-based, 7-metabolite signature for the early diagnosis of Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 1157–1173. [Google Scholar] [PubMed]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Goto, T.; Uchida, M.; Nishimura, K.; Ando, M.; Kobayashi, N.; Goto, J. Presence of protein-bound unconjugated bile acids in the cytoplasmic fraction of rat brain. J. Lipid Res. 2004, 45, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Sato, Y.; Nagata, M.; Goto, T.; Goto, J. Bioconversion of 3β-hydroxy-5-cholenoic acid into chenodeoxycholic acid by rat brain enzyme systems. J. Lipid Res. 2004, 45, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Denis, S.; Faust, P.L.; Wanders, R.J. Bile acids: The role of peroxisomes. J. Lipid Res. 2009, 50, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, R.M.; Borralho, P.M.; Castro, R.E.; Solá, S.; Steer, C.J.; Rodrigues, C.M. Tauroursodeoxycholic acid modulates p53-mediated apoptosis in Alzheimer’s disease mutant neuroblastoma cells. J. Neurochem. 2006, 98, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.M.; Rodrigues, C.M.; Zhao, L.R.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid improves the survival and function of nigral transplants in a rat model of Parkinson’s disease. Cell Transplant. 2002, 11, 195–205. [Google Scholar] [PubMed]
- Yanguas-Casás, N.; Barreda-Manso, M.A.; Nieto-Sampedro, M.; Romero-Ramírez, L. Tauroursodeoxycholic acid reduces glial cell activation in an animal model of acute neuroinflammation. J. Neuroinflamm. 2014, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Chevallier, O.P.; Roberts, D.; Hölscher, C.; Elliott, C.T.; Green, B.D. Investigation of the human brain metabolome to identify potential markers for early diagnosis and therapeutic targets of Alzheimer’s disease. Anal. Chem. 2013, 85, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Holscher, C.; McClean, P.L.; Elliott, C.T.; Green, B. 1H NMR metabolomics investigation of an Alzheimer’s disease (AD) mouse model pinpoints important biochemical disturbances in brain and plasma. Metabolomics 2013, 9, 974–983. [Google Scholar] [CrossRef]
- Graham, S.F.; Holscher, C.; Green, B.D. Metabolic signatures of human Alzheimer’s disease (AD): 1H-NMR analysis of the polar metabolome of post-mortem brain tissue. Metabolomics 2014, 10, 744–753. [Google Scholar] [CrossRef]
- Graham, S.F.; Chevallier, O.P.; Elliott, C.T.; Hölscher, C.; Johnston, J.; McGuinness, B.; Kehoe, P.G.; Passmore, A.P.; Green, B.D. Untargeted metabolomic analysis of human plasma indicates differentially affected polyamine and l-arginine metabolism in mild cognitive impairment subjects converting to Alzheimer’s disease. PLoS ONE 2015, 10, e0119452. [Google Scholar]
- Proitsi, P.; Kim, M.; Whiley, L.; Pritchard, M.; Leung, R.; Soininen, H.; Kloszewska, I.; Mecocci, P.; Tsolaki, M.; Vellas, B.; et al. Plasma lipidomics analysis finds long chain cholesteryl esters to be associated with Alzheimer’s disease. Transl. Psychiatry 2015, 5, e494. [Google Scholar] [CrossRef] [PubMed]
- Fraser, T.; Tayler, H.; Love, S. Fatty acid composition of frontal, temporal and parietal neocortex in the normal human brain and in Alzheimer’s disease. Neurochem. Res. 2010, 35, 503–513. [Google Scholar] [CrossRef]
- Nasaruddin, M.L.; Hölscher, C.; Kehoe, P.; Graham, S.F.; Green, B.D. Wide-ranging alterations in the brain fatty acid complement of subjects with late Alzheimer’s disease as detected by GC-MS. Am. J. Transl. Res. 2016, 8, 154–165. [Google Scholar] [PubMed]
- Pan, X.; Nasaruddin, M.B.; Elliott, C.T.; McGuinness, B.; Passmore, A.P.; Kehoe, P.G.; Hölscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Alzheimer’s disease-like pathology has transient effects on the brain and blood metabolome. Neurobiol. Aging 2016, 38, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Weill-Engerer, S.; David, J.P.; Sazdovitch, V.; Liere, P.; Eychenne, B.; Pianos, A.; Schumacher, M.; Delacourte, A.; Baulieu, E.E.; Akwa, Y. Neurosteroid quantification in human brain regions: Comparison between Alzheimer’s and nondemented patients. J. Clin. Endocrinol. Metab. 2002, 87, 5138–5143. [Google Scholar] [CrossRef] [PubMed]
- Volianskis, A.; Kostner, R.; Molgaard, M.; Hass, S.; Jensen, M.S. Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1 Delta E9-deleted transgenic mice model of beta-amyloidosis. Neurobiol. Aging 2010, 31, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Callaghan, D.; Wodzinska, J.; Xu, J.; Premyslova, M.; Liu, Q.-Y.; Connelly, J.; Zhang, W. Biochemical and behavioral characterization of the double transgenic mouse model (APPswe/PS1dE9) of Alzheimer’s disease. Neurosci. Bull. 2011, 27, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Glossmann, H. The Bile Acid Metabolome in Humans and Rodents, Biocrates Commentary. Available online: http://www.biocrates.com/images/Glossmann2015_HumansANDRodents.pdf (accessed on 12 January 2016).
- Jones, M.L.; Martoni, C.J.; Ganopolsky, J.G.; Labbé, A.; Prakash, S. The human microbiome and bile acid metabolism: Dysbiosis, dysmetabolism, disease and intervention. Expert Opin. Biol. Ther. 2014, 14, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef] [PubMed]
- Hackshaw, A. Small studies: Strengths and limitations. Eur. Respir. J. 2008, 32, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Axelson, M.; Mörk, B.; Sjövall, J. Occurrence of 3β-hydroxy-5-cholestenoic acid, 3β,7α-dihydroxy-5-cholestenoic acid, and 7α-hydroxy-3-oxo-4-cholestenoic acid as normal constituents in human blood. J. Lipid Res. 1988, 29, 629–641. [Google Scholar] [PubMed]
- Nagata, K.; Takakura, K.; Asano, T.; Seyama, Y.; Hirota, H.; Shigematsu, N.; Shima, I.; Kasama, T.; Shimizu, T. Identification of 7α-hydroxy-3-oxo-4-cholestenoic acid in chronic subdural hematoma. Biochim. Biophys. Acta 1992, 1126, 229–236. [Google Scholar] [CrossRef]
- Nagata, K.; Seyama, Y.; Shimizu, T. Changes in the level of 7α-hydroxy-3-oxo-4-cholestenoic acid in cerebrospinal fluid after subarachnoid hemorrhage. Neurol. Med. Chir. 1995, 35, 294–297. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of alzheimer’s disease: Report of the NINCDS-ADRDA work group under the auspices of department of healthand human services task force on Alzheimer’s disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Enot, D.P.; Dallmann, G.; Korner, L.; Forcher, V.; Enoh, P.; Koal, T.; Keller, M.; Deigner, H.P. Complexity and pitfalls of mass spectrometry-based targeted metabolomics in brain research. Anal. Biochem. 2010, 406, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0-making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Bile Acid | Control (n = 10) | AD (n = 10) | p-Value | ROC |
|---|---|---|---|---|
| CA | 947 ± 483 | 156 ± 74 | 0.03 | 0.77 |
| CDCA | 2781 ± 2329 | 312.2 ± 147 | 0.14 | 0.62 |
| DCA | 938 ± 375 | 638 ± 212 | 0.28 | 0.55 |
| GCA | 281 ± 76 | 364 ± 67 | 0.14 | 0.63 |
| GCDCA | 1218 ± 261 | 1118 ± 162 | 0.47 | 0.56 |
| GDCA | 625 ± 125 | 831 ± 199 | 0.31 | 0.58 |
| GLCA | 42 ± 12 | 42 ± 10 | 0.39 | 0.53 |
| GLCAS | 484 ± 99 | 687 ± 126 | 0.14 | 0.67 |
| GUDCA | 141 ± 40 | 170 ± 84 | 0.41 | 0.54 |
| HDCA | <LOD | <LOD | - | - |
| LCA | 63 ± 19 | 44 ± 10 | 0.27 | 0.63 |
| α-MCA | <LOD | <LOD | - | - |
| β-MCA | <LLOQ | <LLOQ | 0.08 | - |
| Ω-MCA | 39 ± 18 | 15 ± 9 | 0.25 | 0.71 |
| TCA | 39 ± 12 | 87 ± 25 | 0.09 | 0.72 |
| TCDCA | 147 ± 39 | 184 ± 56 | 0.38 | 0.51 |
| TDCA | 60 ± 13 | 106 ± 33 | 0.21 | 0.64 |
| TLCA | <LLOQ | <LLOQ | - | - |
| TLCAS | <LLOQ | <LLOQ | - | - |
| TMCA (α + β) | <LLOQ | <LLOQ | - | - |
| TUDCA | <LLOQ | <LLOQ | 0.40 | - |
| UDCA | 206 ± 125 | 101 ± 25 | 0.31 | 0.52 |
| Bile Acid | 6 Months | 12 Months | ||||||
|---|---|---|---|---|---|---|---|---|
| Control (n = 5) | APP/PS1 (n = 5) | p-Value | ROC | Control (n = 5) | APP/PS1 (n = 5) | p-Value | ROC | |
| CA | 1811 ± 368 | 10510 ± 4498 | 0.05 | 0.80 | 2934 ± 1247 | 2099 ± 763 | 0.45 | 0.52 |
| CDCA | 142 ± 44 | 392 ± 189 | 0.18 | 0.68 | 300 ± 66 | 197 ± 90 | 0.17 | 0.68 |
| DCA | 970 ± 331 | 3799 ± 2247 | 0.08 | 0.80 | 1569 ± 431 | 1642 ± 422 | 0.37 | 0.52 |
| GCA | <LLOQ | <LLOQ | 0.19 | - | <LLOQ | <LLOQ | - | - |
| GCDCA | <LOD | <LOD | - | - | <LOD | <LOD | - | - |
| GDCA | <LOD | <LOD | - | - | <LOD | <LOD | - | - |
| GLCA | <LOD | <LOD | - | - | <LOD | <LOD | - | - |
| GLCAS | <LLOQ | <LLOQ | - | - | <LLOQ | <LLOQ | - | - |
| GUDCA | <LLOQ | <LLOQ | - | - | <LLOQ | <LLOQ | - | - |
| HDCA | 609 ± 214 | 405 ± 125 | 0.20 | 0.64 | 1009 ± 226 | 364 ± 192 | 0.04 | 0.87 |
| LCA | 40 ± 10 | 64 ± 32 | 0.32 | 0.56 | 77 ± 15 | 88 ± 32 | 0.48 | 0.52 |
| α-MCA | 88 ± 50 | 211 ± 103 | 0.13 | 0.65 | 391 ± 176 | 136 ± 73 | 0.37 | 0.72 |
| β-MCA | 3132 ± 977 | 8780 ± 3781 | 0.10 | 0.76 | 4271 ± 1706 | 4716 ± 1258 | 0.30 | 0.60 |
| Ω-MCA | 3068 ± 1035 | 5246 ± 2189 | 0.24 | 0.68 | 2368 ± 1213 | 1235 ± 332 | 0.42 | 0.52 |
| TCA | 865 ± 251 | 9473 ± 9007 | 0.36 | 0.60 | 1258 ± 244 | 1051 ± 331 | 0.31 | 0.53 |
| TCDCA | 52 ± 10 | 610 ± 575 | 0.36 | 0.58 | 87 ± 15 | 68 ± 17 | 0.18 | 0.70 |
| TDCA | 143 ± 40 | 912 ± 822 | 0.34 | 0.52 | 159 ± 11 | 188 ± 43 | 0.40 | 0.60 |
| TLCA | <LLOQ | <LLOQ | - | - | <LLOQ | <LLOQ | - | - |
| TLCAS | <LLOQ | <LLOQ | - | - | <LLOQ | <LLOQ | - | - |
| TMCA (α + β) | 1529 ± 322 | 5406 ± 4260 | 0.38 | 0.52 | 1326 ± 318 | 1408 ± 384 | 0.44 | 0.52 |
| TUDCA | 347 ± 72 | 1723 ± 1582 | 0.42 | 0.68 | 990 ± 437 | 513 ± 221 | 0.22 | 0.76 |
| UDCA | 363 ± 150 | 1348 ± 784 | 0.10 | 0.72 | 693 ± 179 | 838 ± 287 | 0.26 | 0.52 |
| Bile Acid | Control (n = 10) | AD (n = 10) | p-Value |
|---|---|---|---|
| CA | 0.16 ± 0.04 | 0.24 ± 0.1 | 0.39 |
| CDCA | 0.33 ± 0.11 | 0.65 ± 0.32 | 0.41 |
| DCA | 0.37 ± 0.11 | 1.84 ± 1.39 | 0.47 |
| GCA | 0.13 ± 0.03 | 0.10 ± 0.02 | 0.25 |
| GCDCA | 0.19 ± 0.08 | 0.14 ± 0.05 | 0.49 |
| GDCA | 0.06 ± 0.02 | 0.10 ± 0.04 | 0.46 |
| GLCA | <LOD | <LOD | - |
| GLCAS | <LLOQ | <LLOQ | - |
| GUDCA | <LLOQ | <LLOQ | - |
| HDCA | <LLOQ | <LLOQ | - |
| LCA | 0.06 ± 0.02 | 0.05 ± 0.01 | 0.44 |
| α-MCA | <LOD | <LOD | - |
| β-MCA | <LOD | <LOD | - |
| Ω-MCA | <LLOQ | <LLOQ | - |
| TCA | 0.06 ± 0.02 | 0.01 ± 0.006 | 0.01 |
| TCDCA | 0.11 ± 0.03 | 0.04 ± 0.01 | 0.07 |
| TDCA | <LLOQ | <LLOQ | - |
| TLCA | <LOD | <LOD | - |
| TLCAS | <LOD | <LOD | - |
| TMCA (α + β) | <LOD | <LOD | - |
| TUDCA | <LOD | <LOD | - |
| UDCA | 0.06 ± 0.01 | 0.16 ± 0.09 | 0.31 |
| Bile Acid | 6 Months | 12 Months | ||||
|---|---|---|---|---|---|---|
| Control (n = 5) | APP/PS1 (n = 5) | p-value | Control (n = 5) | APP/PS1 (n = 5) | p-Value | |
| CA | 0.09 ± 0.01 | 0.10 ± 0.02 | 0.47 | 0.28 ± 0.08 | 0.08 ± 0.02 | 0.02 |
| CDCA | <LOD | <LOD | - | <LOD | <LOD | - |
| DCA | 0.07 ± 0.01 | 0.09 ± 0.02 | 0.35 | 0.05 ± 0.003 | 0.04 ± 0.01 | 0.17 |
| GCA | <LOD | <LOD | - | <LOD | <LOD | - |
| GCDCA | <LLOQ | <LLOQ | - | <LLOQ | <LLOQ | - |
| GDCA | <LOD | <LOD | - | <LOD | <LOD | - |
| GLCA | <LOD | <LOD | - | <LOD | <LOD | - |
| GLCAS | <LOD | <LOD | - | <LOD | <LOD | - |
| GUDCA | <LOD | <LOD | - | <LOD | <LOD | - |
| HDCA | <LOD | <LOD | - | <LOD | <LOD | - |
| LCA | 0.01 ± 0.01 | 0.04 ± 0.01 | 0.05 | 0.04 ± 0.01 | 0.05 ± 0.02 | 0.47 |
| α-MCA | <LOD | <LOD | - | <LOD | <LOD | - |
| β-MCA | 0.08 ± 0.02 | 0.07 ± 0.02 | 0.37 | 0.24 ± 0.06 | 0.08 ± 0.04 | 0.02 |
| Ω-MCA | 0.05 ± 0.01 | 0.03 ± 0.01 | 0.22 | 0.08 ± 0.02 | 0.03 ± 0.01 | 0.05 |
| TCA | 0.10 ± 0.03 | 0.06 ± 0.03 | 0.11 | 0.16 ± 0.07 | 0.04 ± 0.02 | 0.04 |
| TCDCA | <LOD | <LOD | - | <LOD | <LOD | - |
| TDCA | <LOD | <LOD | - | <LOD | <LOD | - |
| TLCA | <LOD | <LOD | - | <LOD | <LOD | - |
| TLCAS | <LOD | <LOD | - | <LOD | <LOD | - |
| TMCA (α + β) | 0.14 ± 0.02 | 0.08 ± 0.03 | 0.05 | 0.25 ± 0.07 | 0.04 ± 0.01 | 0.002 |
| TUDCA | 0.02 ± 0.01 | 0.02 ± 0.002 | 0.33 | 0.03 ± 0.01 | 0.01 ± 0.001 | 0.02 |
| UDCA | <LOD | <LOD | - | <LOD | <LOD | - |
| Sample type | Demographics | AD (n = 10) | Control (n = 10) |
|---|---|---|---|
| Human Brain | Age (years: mean (sd)) | 76.2 (2.3) | 74.5 (3.5) |
| Range (min-max) | 71–79 | 68–80 | |
| Gender F:M | 5:5 | 4:6 | |
| Human Plasma | Age (years: mean (sd)) | 76.3 (5.6) | 77.6 (7.5) |
| Range (min-max) | 70–88 | 66–87 | |
| MMSE (mean (sd)) | 21.9 (4.8) | 29.3 (0.8) | |
| Gender F:M | 6:4 | 5:5 |
| Bile Acid | Abbreviation | Empirical Formula | Molecular Mass | IUPAC Name | |
|---|---|---|---|---|---|
| 1 | Cholic acid | CA | C24H40O5 | 408.57 | (3α,5β,7α,12α)-3,7,12-Trihydroxycholan-24-oic acid |
| 2 | Chenodeoxycholic acid | CDCA | C24H40O4 | 392.57 | (3α,5β,7α,8ξ)-3,7-Dihydroxycholan-24-oic acid |
| 3 | Deoxycholic acid | DCA | C24H40O4 | 392.57 | (3α,5β,12α)-3,12-Dihydroxycholan-24-oic acid |
| 4 | Glycocholic acid | GCA | C26H43NO6 | 465.62 | (3α,5β,7α,8ξ,12α,20R,24Z)-3,7,12,24-Tetrahydroxycholan-24-ylidene]glycine |
| 5 | Glycochenodeoxycholic acid | GCDCA | C26H43NO5 | 449.62 | (3α,5β,7α,8ξ,20R,24Z)-3,7,24-Trihydroxycholan-24-ylidene]glycine |
| 6 | Glycodeoxycholic acid | GDCA | C26H43NO5 | 449.62 | (3α,5β,12α,20R,24Z)-3,12,24-Trihydroxycholan-24-ylidene]glycine |
| 7 | Glycolithocholic acid | GLCA | C26H43NO4 | 433.62 | 3α-hydroxy-5β–cholan-24-oylglycine |
| 8 | Glycolithocholic acid sulphate | GLCAS | C26H42NO7S | 512.27 | 3α-hydroxy-5β-cholan-24-oyl)-glycine 3-sulphate |
| 9 | Glycoursodeoxycholic acid | GUDCA | C26H43NO5 | 449.31 | 3α,7β-dihydroxy-5β–cholan-24-oylglycine |
| 10 | Hyodeoxycholic acid | HDCA | C24H40O4 | 392.57 | (3α,5β,6α)-3,6-Dihydroxycholan-24-oic acid |
| 11 | Lithocholic acid | LCA | C24H40O3 | 376.57 | (3α,5β)-3-Hydroxycholan-24-oic acid |
| 12 | α-muricholic acid | α-MCA | C24H40O5 | 408.57 | (3α,5β,6β,7α)-3,6,7-Trihydroxycholan-24-oic acid |
| 13 | β-muricholic acid | β-MCA | C24H40O5 | 408.57 | (3α,5β,6β,7β)-3,6,7-Trihydroxycholan-24-oic acid |
| 14 | Ω-muricholic acid | Ω-MCA | C24H40O5 | 408.57 | (3α,5β,6α,7β)-3,6,7-Trihydroxycholan-24-oic acid |
| 15 | Taurocholic acid | TCA | C26H45NO7S | 515.70 | (3α,5β,7α,8ξ,12α,20R,24Z)-3,7,12-Trihydroxy-N-(2-sulphoethyl)cholan-24-imidic acid |
| 16 | Taurochenodesoxycholic acid | TCDCA | C26H45NO6S | 499.70 | 2-{[(3α,5β,7α,8ξ,9ξ,14ξ)-3,7-Dihydroxy-24-oxocholan-24-yl]amino}ethanesulphonic acid |
| 17 | Taurodeoxycholic acid | TDCA | C26H45NO6S | 499.70 | α,12α-dihydroxy-5β–cholan-24-oyltaurine |
| 18 | Taurolithocholic acid | TLCA | C26H45NO5S | 483.70 | (3α,5β,20R,24Z)-3-Hydroxy-N-(2-sulphoethyl)cholan-24-imidic acid |
| 19 | Taurolithocholic acid sulphate | TLCAS | C26H45NO8S2 | 563.76 | (3α,5β,20R,24Z)-N-(2-Sulphoethyl)-3-(sulphooxy)cholan-24-imidic acid |
| 20 | Tauromuricholic acid (α and β) | TMCA (α + β) | C26H45NO7S | 515.70 | 3α,6β,7α/β -trihydroxy-5β–cholan-24-oyltaurine |
| 21 | Tauroursodeoxycholic acid | TUDCA | C26H45NO6S | 499.70 | 2-{[(3α,5β,7β)-3,7-Dihydroxy-24-oxocholan-24-yl]amino}ethanesulphonic acid |
| 22 | Ursodeoxycholic acid | UDCA | C24H40O4 | 392.57 | (3α,5β,7β)-3,7-Dihydroxycholan-24-oic acid |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Elliott, C.T.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Hölscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites 2017, 7, 28. https://doi.org/10.3390/metabo7020028
Pan X, Elliott CT, McGuinness B, Passmore P, Kehoe PG, Hölscher C, McClean PL, Graham SF, Green BD. Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites. 2017; 7(2):28. https://doi.org/10.3390/metabo7020028
Chicago/Turabian StylePan, Xiaobei, Christopher T. Elliott, Bernadette McGuinness, Peter Passmore, Patrick G. Kehoe, Christian Hölscher, Paula L. McClean, Stewart F. Graham, and Brian D. Green. 2017. "Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease" Metabolites 7, no. 2: 28. https://doi.org/10.3390/metabo7020028
APA StylePan, X., Elliott, C. T., McGuinness, B., Passmore, P., Kehoe, P. G., Hölscher, C., McClean, P. L., Graham, S. F., & Green, B. D. (2017). Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites, 7(2), 28. https://doi.org/10.3390/metabo7020028