1. Introduction
In recent years, the systems biology of metabolism has moved more and more from classical metabolic network study towards the study of growth as a result of an optimized cellular economy. This idea of studying growth strategies using resource allocation models has been initiated by Molenaar et al. [
1] in 2009. In their article, Molenaar et al. used a small dynamic model of a self-replicating system to explain how overflow metabolism arises by means of tradeoffs between different growth strategies. Further on, Goelzer et al. [
2] introduced resource balance analysis (RBA), as a means of predicting the cell composition of bacteria in a specific (constant) environment through a convex optimization problem that includes the bioenergetic cost of producing the enzymes required in a pathway. As a similar approach, Palsson and colleagues introduced the idea of an integrated model of metabolism and gene expression (ME model) as a means to explore the relationship between genotype and phenotype using biochemical representations of transcription and translation processes [
3,
4]. Their research group then continued with an ME model of
Escherichia coli [
5]. With the COBRAme package [
6], a computational framework for building and manipulating ME models is provided. Also experimental studies focused on relating absolute protein abundances to how metabolic pathways balance production costs and activity requirements [
7].
These formalisms have then been taken a step forward, towards understanding how resources are distributed in a dynamically changing environment by means of a dynamic enzyme-cost flux balance analysis (deFBA) [
8] and conditional flux balance analysis (cFBA) [
9]. This has then been taken to the genome scale by studying the optimal glycogen and metabolite partitioning dynamics under a day-night cycle in a cyanobacterium using a dynamic resource allocation model [
10].
Such dynamic resource allocation models have a wide area of application. One such an example is the study of microorganisms growing in industry-scale bioreactors. There, the organism has to balance resources not only in order to grow optimally, but also in order to withstand transitions through local heterogeneities of the reactor. The ability to take such transitions into account within metabolism has been shown to be crucial for survival [
11]. Moreover, an extension of the deFBA formalism has been developed in order to predict the optimal resource allocation in an environment where such uncertainties are present [
12].
Given all these recent developments, we believe that there is a need to establish a protocol for building a metabolic resource allocation model. However, to the best of our knowledge, there exists no generic guideline that details how to proceed in the construction of large-scale metabolic resource allocation models, together with possible sources of the relevant parameters. Moreover, there exists so far no specification for defining and exchanging these models that would be similarly useful as the current SBML standard for kinetic and metabolic flux balance models.
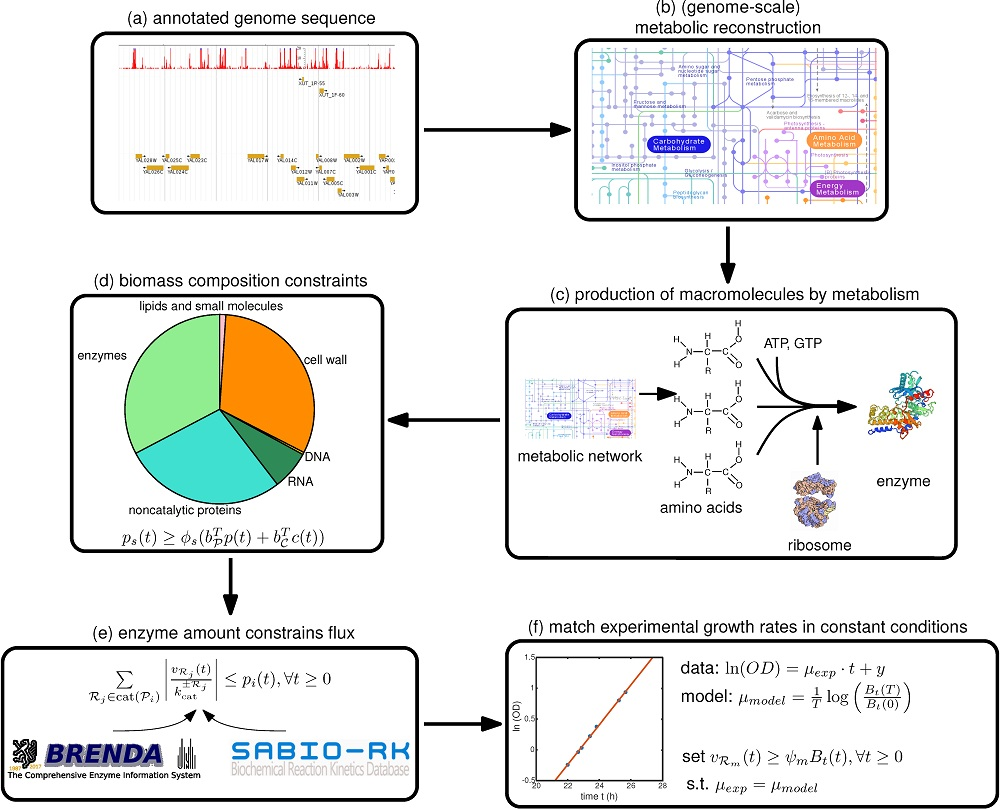
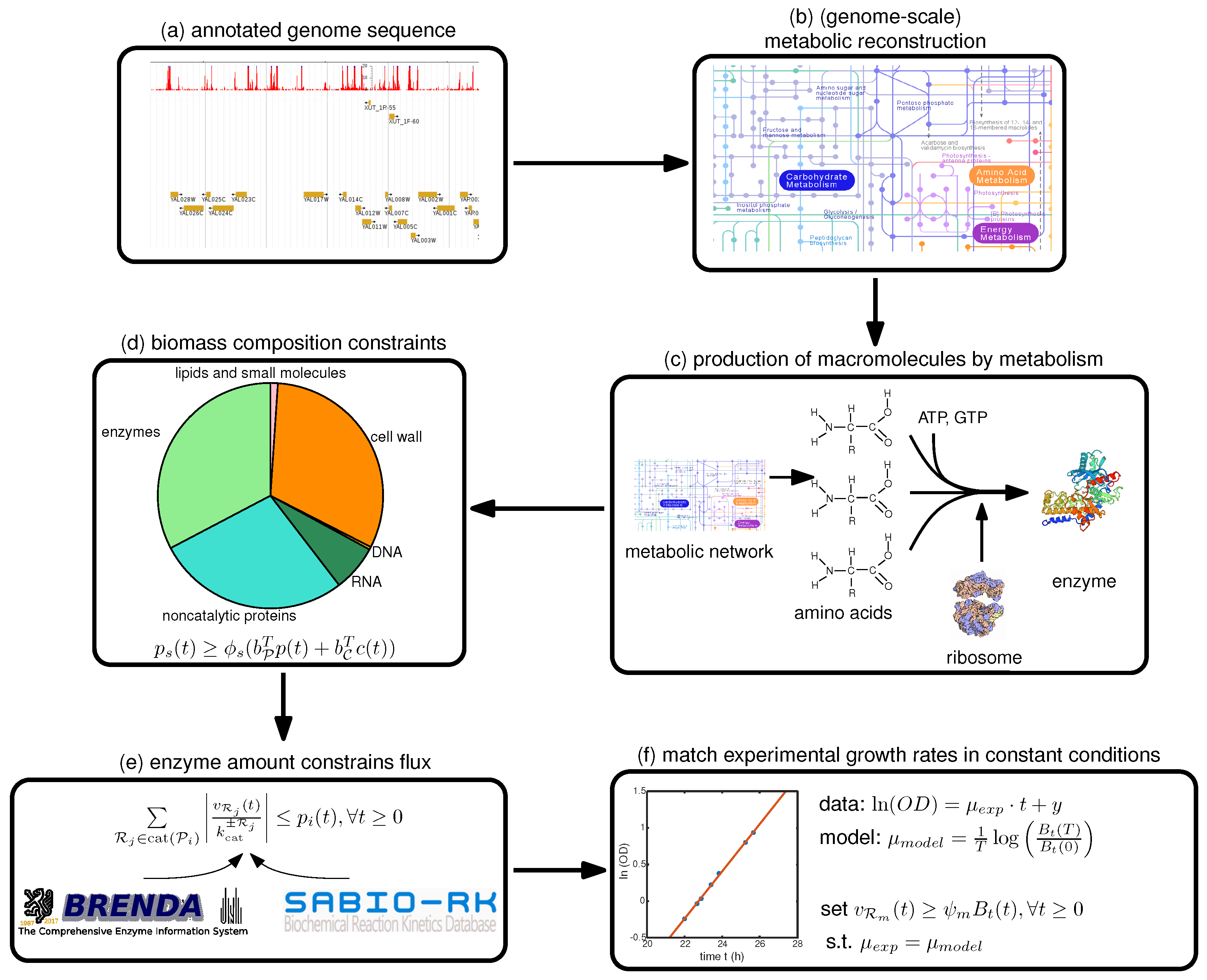
Therefore, we focus in this paper on a step-by-step guide towards constructing such a model, summarized in
Figure 1, with a focus on the deFBA formalism which is described in the Methods section. Note however, that these guidelines can be used as well for building cFBA, RBA and ME models. We detail here all the necessary information as well as which databases may be used to retrieve it (
Table 1). To facilitate exchange among researchers, we furthermore propose a new SBML specification, called resource allocation modeling (RAM) [
13]. This specification allows encoding such models in the SBML format using the Flux Balance Constraints extension [
14].
In addition to this protocol, we provide software in Python 2.7 (implementation can be found at:
https://bitbucket.org/hlindhor/defba-python-package) as well as MATLAB R2016a (implementation is available at
https://github.com/alexandra-m-reimers/deFBA) for reading and writing resource allocation models using our SBML specification as well as for solving deFBA problems. We would like to note that these models are strongly organism-dependent. Therefore, the modeler is still required to decide which key processes are modeled and which level of detail is used for their particular application.
2. Methods
The dynamic enzyme-cost flux balance analysis models a metabolic reaction network coupled with gene expression as a dynamic optimization problem. By assuming the system to be self-optimizing for growth, regulatory features of the network, which are often not known in detail, need not be explicitly included in the model. Instead, the reaction fluxes are used as decision variables for the optimization problem. We present very shortly the mathematical notation of deFBA, so that the reader can understand the problems we face when building these models.
We will use sets of indices to denote submatrices and subvectors. For instance, denotes the submatrix of S corresponding to the rows in the set A and all columns, while denotes the subvector of v with the entries at the indices in . Furthermore, we use to denote the number of elements of a set A.
The model consists of n species divided into four different groups:
the set of external species , present in the environment (e.g., carbon sources, oxygen, nitrogen), with corresponding molar amounts , ,
the set of internal metabolic species acting as precursors for the production of biomass (e.g., ATP, NADH, amino acids), with corresponding molar amounts , ,
the set of storage species , which save energy for later usage (e.g., starch, glycogen), with corresponding molar amounts , ,
the set of macromolecules , which are catalytic enzymes or necessary cellular building blocks, with corresponding molar amounts , ,
with , .
The deFBA model is a dynamic model and hence, all variables described above are considered as functions of time. As in most constraint-based modeling frameworks for metabolism, deFBA assumes that the cell has evolved to maximize its growth in the form of maximizing total biomass at each time point in the simulation period. Thus, we use the
objective weights , which are typically identical to the molecular weights
,
, for all macromolecules
to define the
objective biomass until end-time
, as
Additionally, we define the
total biomass by adding the weight of the storage
The optimization problem is constructed with the assumption that reaction rates (fluxes)
,
, which are also time-dependent, are chosen to maximize the biomass accumulation over the simulation time
given the initial macromolecule amounts
. The objective function is thus constructed as the biomass integral
in which we use the objective biomass Equation (
1).
Note that, although in our formulation storage species are not part of the objective biomass, the deFBA formalism does not strictly prohibit this. This means that, if for the modeled organism the storage should be part of the objective biomass, this can be incorporated. Furthermore, note that we allow some of the objective weights to be zero, in order to account, e.g., for the possibility that the modeled organism secretes enzymes that then catalyze external reactions.
As with the species we differentiate the r reactions into four groups
the set of exchange and external reactions , which transport matter between the cell and the environment or convert external species into each other, with corresponding fluxes , ,
the set of internal metabolic reactions , which convert internal metabolites into each other, with corresponding fluxes , ,
the set of storage reactions , which convert between internal metabolites and storage, with corresponding fluxes , ,
the set of biomass reactions , which synthesize macromolecules from internal metabolite precursors, with corresponding fluxes , ,
where , and the set of all reactions is given by .
We note that in deFBA models each reaction is producing a biomass component, as opposed to regular FBA models, which only maximize the flux through a single biomass producing reaction.
The differential equations describing the dynamics of the species are given by the stoichiometric matrix
as
for all
, where the entries
give the stoichiometry of species
i in reaction
j.
The complexity of the problem is reduced using a quasi-steady-state approximation for the internal metabolites as
Furthermore, flux constraints which are independent of enzymatic capacity can be added as
In flux balance analysis (FBA) [
21,
22], where only the part of the system corresponding to internal and exchange reactions is modeled and a static biomass objective function is maximized, these box constraints are necessary to limit the growth yield, defined as the flux through the biomass reaction. For our application, the limiting factor for the growth rate is the capacity of the enzymes to catalyze the reactions, depending on the
catalytic constants . Individual enzymes may catalyze multiple reactions. Hence, we denote the set of reactions catalyzed by the enzyme
as
and constrain the reactions fluxes via
with the forward (backward) constant
(
),
. Similarly, the amount of ribosome constrains the total rate of protein synthesis in the model. All these constraints can be formulated linearly as
with the
capacity matrix containing the catalytic constants and the
filter matrix containing exactly one non-zero entry per row. An example of how to construct the matrices
and
for the model introduced in
Section 8 can be found in the Supplements. The
enzyme capacity constraint Inequality (
7) must be satisfied at all times. Assuming any pathway from nutrients to biomass contains at least one reaction limited by an enzyme, the rate of this reaction will be limiting and thus the growth rate will be finite at all times.
In addition to enzymes and ribosomes, deFBA models also include noncatalytic biomass. These are macromolecules of the cell that fulfill no immediate catalytic activity, such as the cell wall or the membrane, but are nevertheless crucial for reproduction and their synthesis consumes cellular resources. To model this, we impose a constraint to enforce the production of a certain noncatalytic biomass component in a proportional way with the catalytic biomass. We call these species
quota compounds. As an example, consider a quota macromolecule
and assume it must make up 20% of the total biomass
at any time point
. We express this as
with
. We call the according matrix formulation the
biomass composition constraint and write
An example of
for the model in
Section 8 can be found in the Supplements.
Finally, since deFBA models do not include all resource and energy consuming processes in the cell, an ATP-maintenance reaction may be used to tune the model-derived growth rate and represent additional unmodeled energy sinks. An ATP-maintenance reaction hydrolyzes ATP as
These reactions are typically enforced proportionally to the total biomass. Thus we assign each maintenance reaction
a maintenance coefficient
,
, and write
An example of
for the model in
Section 8 can be found in the Supplements.
We do not include the maintenance reactions as an individual class of reactions, as we are usually only handling very few of them in comparison to other reactions. The maintenance reactions will thus typically be a subset of the metabolic reaction set .
To formulate the dynamic optimization problem we need to choose initial conditions for the external species
, storage species
, and the macromolecules
. In many cases, one can assume that cells are adapted to achieve maximum growth rate in a certain medium in which they have been cultured before the start of the process modeled by deFBA. To obtain the biomass composition in these cases, a good strategy is to solve an RBA problem [
2] with extracellular species amounts
based on the preculture medium, yielding storage and macromolecule amounts
and
for optimal growth in this medium. The initial values are then set as
The metabolites
operate in quasi steady-state (see Equation (14)) and thus do not need initial values. The complete deFBA problem then reads
This dynamic optimization problem can be solved by discretizing time using a collocation method [
8]. This way the problem is cast into a linear program (LP), which can be solved using standard commercial solvers such as CPLEX or Gurobi or open source solvers such as cvxopt [
23] or the more numerically stable SoPlex [
24,
25,
26]. The linearity of the problem is given by modeling molar amounts of species instead of concentrations, see [
8] for further details. With respect to the computational and numerical details of solving such problems, we refer the reader to [
8], and to [
10] for a large scale example.
6. Assigning Reaction Turnover Rates
Turnover rates are necessary parameters in a resource allocation model. They are involved in the capacity constraints on reaction fluxes using the amount of their catalyzing enzymes.
Such turnover rates can be derived from experimental data as explained in [
57]. Alternatively, a recent study has shown that turnover rates reported in online databases are a good enough approximation of in vivo turnover rates [
58].
The two main databases for retrieving turnover rates are BRENDA [
36] and SABIO-RK [
37]. While BRENDA stores both manually curated as well as text mining data, SABIO-RK only offers data that was either manually extracted from the literature or directly submitted by experimenters. As a result, BRENDA offers a larger amount of turnover rates than SABIO-RK. On the other hand, the text mining entries may not have the same quality as the manually curated ones and the incorporation of these values in resource allocation models should be done with care and if possible these values should be manually checked. Both databases have automated retrieval options.
Some simple rules of thumb for retrieving turnover rates from these databases are that one should filter for wild type, non-recombinant values, and, if possible, should make sure that the measurements were done at (nearly) physiological pH. This typically narrows down the results significantly such that alternatives can be investigated or a median of the remaining values can be used, in a similar fashion as explained below.
Although large amounts of biochemical data are now available, usually not all turnover rates for the organism of interest can be found. We recommend that in this case, if turnover rates for a given enzyme from other organisms are found, that these should be used. Moreover, it is important to perform a sensitivity analysis to check the influence of these unknown parameters on the results.
The question then arises: which of the available other organism turnover rates should be used? Should it be a mean or a median of all found turnover rates for the respective enzyme, or the turnover rate from the organism that has the most sequence similarity with the target organism within that protein?
To answer this question, we have automatically retrieved wild type turnover rates from the BRENDA database of all enzymes for three organisms:
Saccharomyces cerevisiae,
Escherichia coli, and
Bacillus subtilis. In a second iteration, we retrieved turnover rates of all enzymes from all other organisms, excluding the organism of interest, and computed the mean, median, and best sequence match with the organism of interest
on a per enzyme basis. The best sequence match was obtained by computing the alignment score using the Needleman-Wunsch algorithm [
59] with the BLOSUM62 scoring matrix [
60]. We computed the Pearson correlation coefficients between the logarithms of
values from the organism of interest and the logarithms of the mean, median and best sequence match
values obtained from other organisms. Only values corresponding to the same catalyzed reaction were compared. The resulting correlation coefficients are displayed in
Table 2.
We observe that, in the cases we have analyzed, the medians of all turnover rates enzyme-wise is the best approximation for the actual turnover rates in the organism of interest. Moreover, the order of magnitude correlation coefficients are very high and the p-values we get are all in the order of or lower, indicating that indeed these median turnover rates from other organisms are good enough approximations of the real values, if no specific data is available for the organism of interest.
To give an idea of the spread of the turnover rate data, we show in
Figure 2 a plot of the
values in yeast versus the median
values from other organisms.
9. Discussion
Dynamic resource allocation models have emerged in recent years as a means of extending the predictive capabilities of constraint-based models. Such resource allocation models allow investigating how dynamics of the extracellular environment are reflected inside the metabolism in the form of cost-benefit tradeoffs of active pathways. This is achieved by extending FBA models in two directions: (i) accounting for the costs of producing enzymes before these can be used to catalyze their corresponding reactions; and (ii) taking a dynamic perspective where the levels of these enzymes change over time in response to changes in the environment.
Such models have a larger predictive power and can predict complex biological behaviors such as the catabolite repression as demonstrated in [
8]. Moreover, a recent genome-scale dynamic resource allocation study [
10] shows that these models can be used to understand the optimality of glycogen accumulation patterns in cyanobacteria. Such bacteria live in a constantly changing environment governed by the dynamics of sun light availability. By using dynamic resource allocation models, it was possible to develop a manifest hypothesis on the biology of cyanobacteria: that their metabolism is coordinated according to a temporal program that evolved to maximize growth in a diurnal environment, and that the circadian clock is the regulatory mechanism that modulates the transcriptional program of the cell to achieve this metabolic optimum.
Although their large predictive power has already been demonstrated, it is not possible to use resource allocation models for studying all biological processes connected to the metabolism. For instance, such models always assume optimality of growth. A scenario where this may not be an accurate assumption is starvation, when the objective changes more towards maximizing survival rather than growth. Moreover, starvation triggers different kinetics of nutrient uptake, where stochastic effects and substrate affinity may play a role. It is, therefore, no longer sufficient to consider only upper bounds on uptake rates based on enzyme amount and turnover rate, as done in resource allocation models.
Another limitation of such models may also lie in the fact that, so far, they do not incorporate any kind of regulatory logic that may also have an impact on the metabolic strategies, in addition to the resource tradeoffs. Some resource allocation models such as ME models indeed go a step beyond modeling translation costs and also model transcription. Others, like deFBA or RBA, include such costs in the form of biomass composition constraints and maintenance.
So far, there exist ME models of
E. coli [
5] and
T. maritima [
4], an RBA model of
B. subtilis [
2,
57], and a deFBA model of
S. elongatus [
10]. Moreover, we can mention software packages for handling ME models [
6] and deFBA models (Python implementation can be found at:
https://bitbucket.org/hlindhor/defba-python-package; MATLAB implementation is available at
https://github.com/alexandra-m-reimers/deFBA). We therefore felt there is a need in the community for a guideline for building such models as well as a means to exchange them without losing information.
Therefore, we have provided in this article a list of ingredients of such models, together with links to the relevant databases where parameters can be found. Every step of this guideline can be automated except for (i) production of enzyme complexes for which the subunit stoichiometries cannot yet be automatically retrieved from online databases; (ii) adjustment of protein quota requirements, which needs a quantitative proteomics dataset; and (iii) maintenance requirements for adjusting growth rate.
With respect to exchanging metabolic resource allocation models, we have provided here a specification that is based on SBML and the flux balance constraints package. This specification covers deFBA and RBA models and we believe that it can be extended to also incorporate ME models.
We believe that our contribution will help extend the use and number of metabolic resource allocation models as well as exchange of these models among researchers.





{kind=link}
{kind=link}
{kind=link}

