Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics



Abstract
:1. Introduction
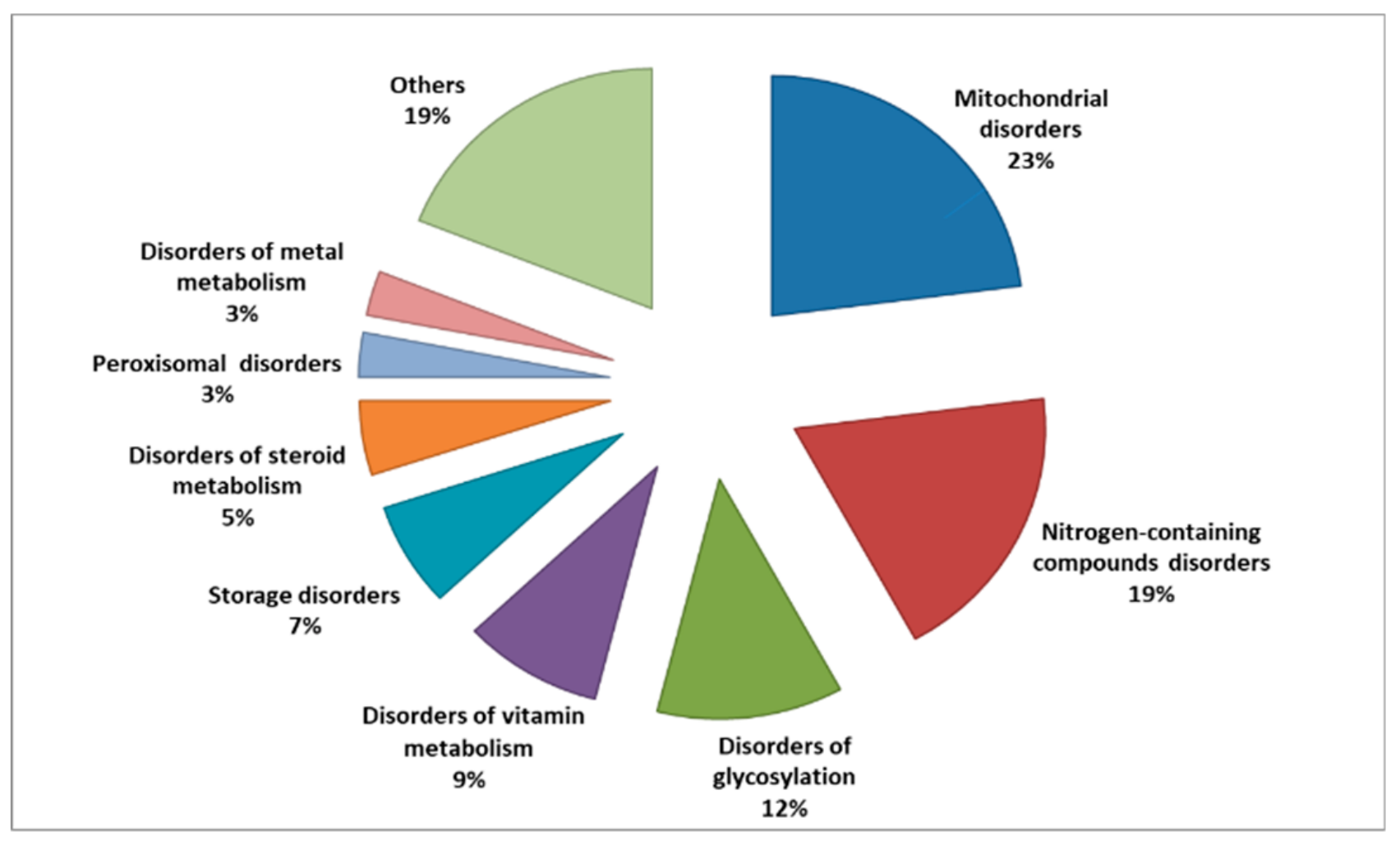
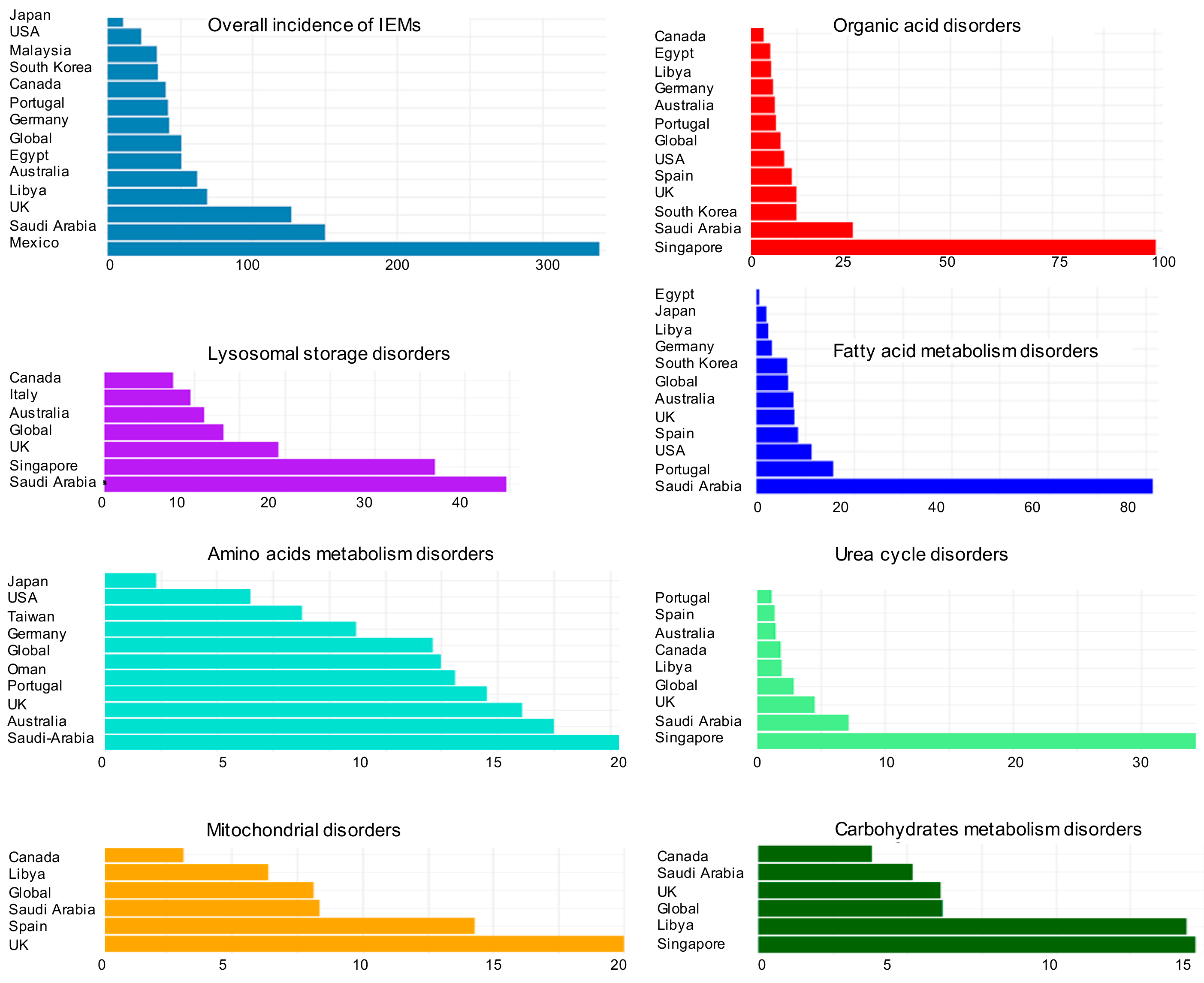
2. Overview of Inborn Errors of Metabolism Diseases
3. Inheritance and Causes
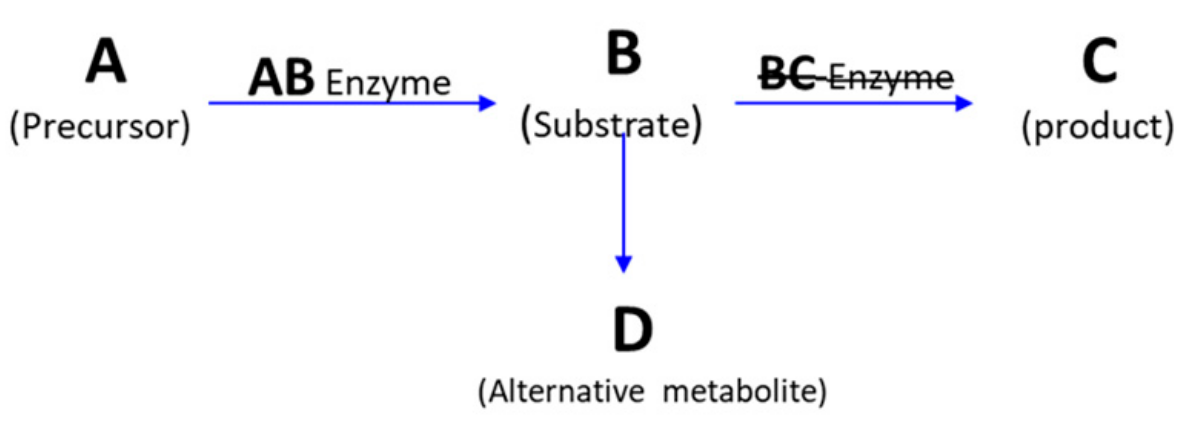
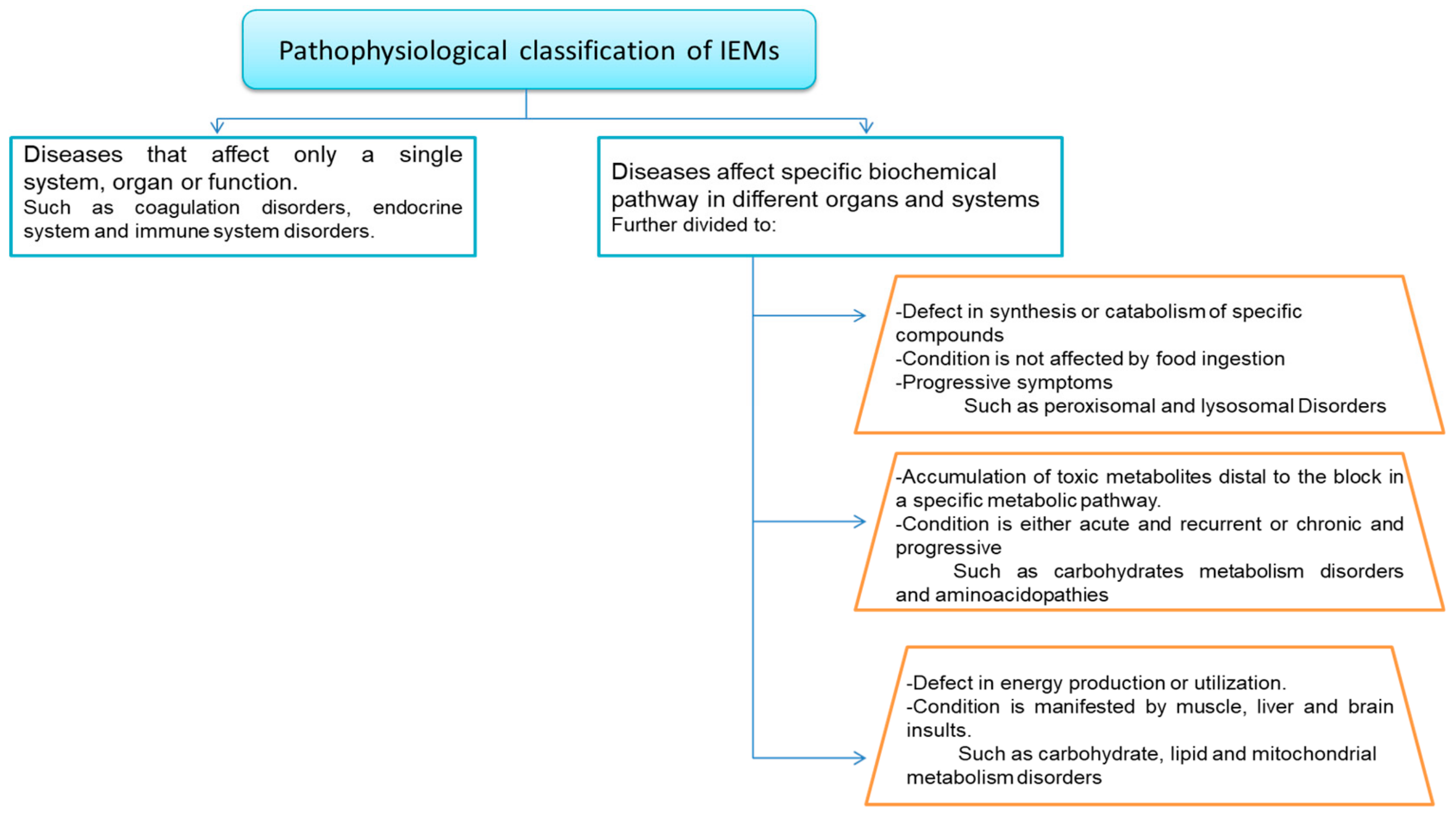
4. Classifications
5. Clinical Presentation and Outcomes
6. Diagnosis and Screening Inborn Errors of Metabolism
7. Metabolomics Technologies Used in Inborn Errors of Metabolism
8. Matrix
9. Methods of Metabolomics Analyses
10. Targeted Metabolomics in the Screening and Diagnosis of Inborn Errors in Metabolism
11. Untargeted Metabolomics in the Screening and Diagnosis of Inborn Errors of Metabolism
12. Lipidomic Studies in Inborn Errors of Metabolism
13. Processing Raw Untargeted Metabolomics Data
14. IEM Screening and Diagnosis Comparing Targeted Versus Untargeted Metabolomics
15. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guthrie, R.; Susi, A. A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [PubMed]
- Williams, R.A.; Mamotte, C.D.; Burnett, J.R. Phenylketonuria: An inborn error of phenylalanine metabolism. Clin. Biochem. Rev. 2008, 29, 31–41. [Google Scholar] [PubMed]
- Matsumoto, I.; Kuhara, T. A new chemical diagnostic method for inborn errors of metabolism by mass spectrometry-rapid, practical, and simultaneous urinary metabolites analysis. Mass Spectrom. Rev. 1996, 15, 43–57. [Google Scholar] [CrossRef]
- Sirrs, S.; Hollak, C.; Merkel, M.; Sechi, A.; Glamuzina, E.; Janssen, M.C.; Mochel, F. The Frequencies of Different Inborn Errors of Metabolism in Adult Metabolic Centres: Report from the SSIEM Adult Metabolic Physicians Group. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2015; Volume 27, pp. 85–91. [Google Scholar]
- Wilson, J.M.; Jungner, Y.G. Principles and practice of mass screening for disease. Bol. Oficina Sanit. Panam. 1968, 65, 281–393. [Google Scholar] [PubMed]
- Therrell, B.L.; Adams, J. Newborn screening in North America. J. Inherit. Metab. Dis. 2007, 30, 447–465. [Google Scholar] [CrossRef] [PubMed]
- Millington, D.S. The Role of Technology in Newborn Screening. N. C. Med. J. 2019, 80, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Kuhara, T. Gas chromatographic-mass spectrometric urinary metabolome analysis to study mutations of inborn errors of metabolism. Mass Spectrom. Rev. 2005, 24, 814–827. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, L.; Mei, H.; Li, X.; Cheng, J.; Cai, Y. Detection of inborn errors of metabolism using GC-MS: Over 3 years of experience in southern China. J. Pediatric Endocrinol. Metab. 2015, 28, 375–380. [Google Scholar] [CrossRef]
- Lehotay, D.C.; Hall, P.; Lepage, J.; Eichhorst, J.C.; Etter, M.L.; Greenberg, C.R. LC-MS/MS progress in newborn screening. Clin. Biochem. 2011, 44, 21–31. [Google Scholar] [CrossRef]
- Wu, J.T. Screening for inborn errors of amino acid metabolism. Ann. Clin. Lab. Sci. 1991, 21, 123–142. [Google Scholar]
- Beutler, E. Galactosemia: Screening and diagnosis. Clin. Biochem. 1991, 24, 293–300. [Google Scholar] [CrossRef]
- Chace, D.H.; Hillman, S.L.; Van Hove, J.L.; Naylor, E.W. Rapid diagnosis of MCAD deficiency: Quantitative analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin. Chem. 1997, 43, 2106–2113. [Google Scholar]
- Ito, T.; van Kuilenburg, A.B.; Bootsma, A.H.; Haasnoot, A.J.; van Cruchten, A.; Wada, Y.; van Gennip, A.H. Rapid screening of high-risk patients for disorders of purine and pyrimidine metabolism using HPLC-electrospray tandem mass spectrometry of liquid urine or urine-soaked filter paper strips. Clin. Chem. 2000, 46, 445–452. [Google Scholar] [PubMed]
- Levy, P.A. Inborn errors of metabolism: Part. 1: Overview. Pediatric Rev. 2009, 30, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, M.; Schwarz, E.; Jensen, M.; Yuzyuk, T.; DeBiase, I.; Randall, H.; Longo, N. Feasibility of newborn screening for guanidinoacetate methyltransferase (GAMT) deficiency. J. Inherit. Metab. Dis. 2014, 37, 231–236. [Google Scholar] [CrossRef]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol. Genet. Metab. 2016, 118, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Bleyle, L.; Huidekoper, H.H.; Vaz, F.M.; Singh, R.; Steiner, R.D.; DeBarber, A.E. Update on newborn dried bloodspot testing for cerebrotendinous xanthomatosis: An available high-throughput liquid-chromatography tandem mass spectrometry method. Mol. Genet. Metab. Rep. 2016, 7, 11–15. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Almannai, M.; Sutton, V.R. Newborn Screening: History, Current Status, and Future Directions. Pediatric Clin. N. Am. 2018, 65, 389–405. [Google Scholar] [CrossRef]
- Kumar, A.B.; Masi, S.; Ghomashchi, F.; Chennamaneni, N.K.; Ito, M.; Scott, C.R.; Turecek, F.; Gelb, M.H.; Spacil, Z. Tandem Mass Spectrometry Has a Larger Analytical Range than Fluorescence Assays of Lysosomal Enzymes: Application to Newborn Screening and Diagnosis of Mucopolysaccharidoses Types II, IVA, and VI. Clin. Chem. 2015, 61, 1363–1371. [Google Scholar] [CrossRef]
- Levy, H.L.; Albers, S. Genetic screening of newborns. Annu. Rev. Genom. Hum. Genet. 2000, 1, 139–177. [Google Scholar] [CrossRef]
- Ghosh, A.; Schlecht, H.; Heptinstall, L.E.; Bassett, J.K.; Cartwright, E.; Bhaskar, S.S.; Urquhart, J.; Broomfield, A.; Morris, A.A.; Jameson, E.; et al. Diagnosing childhood-onset inborn errors of metabolism by next-generation sequencing. Arch. Dis. Child. 2017, 102, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodian, D.L.; Klein, E.; Iyer, R.K.; Wong, W.S.; Kothiyal, P.; Stauffer, D.; Huddleston, K.C.; Gaither, A.D.; Remsburg, I.; Khromykh, A.; et al. Utility of whole-genome sequencing for detection of newborn screening disorders in a population cohort of 1,696 neonates. Genet. Med. 2016, 18, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarini, B.A.; Goldenberg, A.J. Ethical issues with newborn screening in the genomics era. Annu. Rev. Genom. Hum. Genet. 2012, 13, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Andermann, A.; Blancquaert, I.; Beauchamp, S.; Déry, V. Revisiting Wilson and Jungner in the genomic age: A review of screening criteria over the past 40 years. Bull. World Health Organ. 2008, 86, 317–319. [Google Scholar] [CrossRef]
- Knoppers, B.M.; Sénécal, K.; Borry, P.; Avard, D. Whole-genome sequencing in newborn screening programs. Sci. Transl. Med. 2014, 6, 229cm2. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef]
- Argmann, C.A.; Houten, S.M.; Zhu, J.; Schadt, E.E. A Next Generation Multiscale View of Inborn Errors of Metabolism. Cell Metab. 2016, 23, 13–26. [Google Scholar] [CrossRef]
- Kanungo, S.; Patel, D.R.; Neelakantan, M.; Ryali, B. Newborn screening and changing face of inborn errors of metabolism in the United States. Ann. Transl. Med. 2018, 6, 468. [Google Scholar] [CrossRef]
- Holmes, D. Europe plays catch-up on neonatal screening as US skips ahead. Nat. Med. 2012, 18, 1596. [Google Scholar] [CrossRef]
- Grosse, S.D.; Rogowski, W.H.; Ross, L.F.; Cornel, M.C.; Dondorp, W.J.; Khoury, M.J. Population screening for genetic disorders in the 21st century: Evidence, economics, and ethics. Public Health Genom. 2010, 13, 106–115. [Google Scholar] [CrossRef]
- Grosse, S.D.; van Vliet, G. Prevention of intellectual disability through screening for congenital hypothyroidism: How much and at what level? Arch. Dis. Child. 2011, 96, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Morillo, E.; Garcia, B.P.; Menendez, F.V.A. Challenges for Worldwide Harmonization of Newborn Screening Programs. Clin. Chem. 2016, 62, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcken, B.; Wiley, V.; Hammond, J.; Carpenter, K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N. Engl. J. Med. 2003, 348, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Venditti, L.N.; Venditti, C.P.; Berry, G.T.; Kaplan, P.B.; Kaye, E.M.; Glick, H.; Stanley, C.A. Newborn screening by tandem mass spectrometry for medium-chain Acyl-CoA dehydrogenase deficiency: A cost-effectiveness analysis. Pediatricics 2003, 112, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, S.; Green, A.; Preece, M.A.; Burton, H. The incidence of inherited metabolic disorders in the West Midlands, UK. Arch. Dis. Child. 2006, 91, 896–899. [Google Scholar] [CrossRef] [Green Version]
- Freer, D.E.; Ficicioglu, C.; Finegold, D. Newborn screening for galactosemia: A review of 5 years of data and audit of a revised reporting approach. Clin. Chem. 2010, 56, 437–444. [Google Scholar] [CrossRef]
- Bodamer, O.A.; Scott, R.C.; Giugliani, R. Newborn Screening for Pompe Disease. Pediatrics 2017, 140, S4–S13. [Google Scholar] [CrossRef] [Green Version]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Gangoiti, J.A.; Barshop, B.A.; Siuzdak, G. Metabolomics identifies perturbations in human disorders of propionate metabolism. Clin. Chem. 2007, 53, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; van Karnebeek, C.D.M.; Vockley, J.; Blau, N. A proposed nosology of inborn errors of metabolism. Genet. Med. 2019, 21, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Mussap, M.; Zaffanello, M.; Fanos, V. Metabolomics: A challenge for detecting and monitoring inborn errors of metabolism. Ann. Transl. Med. 2018, 6, 338. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Zhou, X.; Chen, X.; Wu, Y.; Liu, C.; Kong, Q. Expanded Newborn Screening for Inborn Errors of Metabolism and Genetic Characteristics in a Chinese Population. Front. Genet. 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzal, R.M.; Lund, A.M.; Skovby, F. The impact of consanguinity on the frequency of inborn errors of metabolism. Mol. Genet. Metab. Rep. 2018, 15, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S. Recognition and diagnostic approach to acute metabolic disorders in the neonatal period. Sudan. J. Paediatrics 2011, 11, 20–28. [Google Scholar]
- Waters, D.; Adeloye, D.; Woolham, D.; Wastnedge, E.; Patel, S.; Rudan, I. Global birth prevalence and mortality from inborn errors of metabolism: A systematic analysis of the evidence. J. Glob. Health 2018, 8, 021102. [Google Scholar] [CrossRef]
- Wertheim-Tysarowska, K.; Gos, M.; Sykut-Cegielska, J.; Bal, J. Genetic analysis in inherited metabolic disorders--from diagnosis to treatment. Own experience, current state of knowledge and perspectives. Dev. Period. Med. 2015, 19, 413–431. [Google Scholar]
- Ezgu, F. Inborn Errors of Metabolism. Adv. Clin. Chem. 2016, 73, 195–250. [Google Scholar]
- Tiwari, S.; Kallianpur, D.; DeSilva, K.A. Communication Impairments in Children with Inborn Errors of Metabolism: A Preliminary Study. Indian J. Psychol. Med. 2017, 39, 146–151. [Google Scholar] [CrossRef]
- Vernon, H.J. Inborn Errors of Metabolism: Advances in Diagnosis and Therapy. JAMA Pediatricic 2015, 169, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Saudubray, J.M.; Garcia-Cazorla, A. Inborn Errors of Metabolism Overview: Pathophysiology, Manifestations, Evaluation, and Management. Pediatricic Clin. N. Am. 2018, 65, 179–208. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K. Inborn errors of metabolism: Challenges and management. Indian J. Clin. Biochem. 2013, 28, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.M. Inborn errors of metabolism: A clinical overview. Sao Paulo Med. J. 1999, 117, 251–265. [Google Scholar] [CrossRef]
- Saudubray, J.M.; Martin, D.; de Lonlay, P.; Touati, G.; Poggi-Travert, F.; Bonnet, D.; Jouvet, P.; Boutron, M.; Slama, A.; Vianey-Saban, C.; et al. Recognition and management of fatty acid oxidation defects: A series of 107 patients. J. Inherit. Metab. Dis. 1999, 22, 488–502. [Google Scholar] [CrossRef]
- Leonard, J.V.; Morris, A.A. Inborn errors of metabolism around time of birth. Lancet 2000, 356, 583–587. [Google Scholar] [CrossRef]
- Colonetti, K.; Roesch, L.F.; Schwartz, I.V.D. The microbiome and inborn errors of metabolism: Why we should look carefully at their interplay? Genet. Mol. Biol. 2018, 41, 515–532. [Google Scholar] [CrossRef] [Green Version]
- Agana, M.; Frueh, J.; Kamboj, M.; Patel, D.R.; Kanungo, S. Common metabolic disorder (inborn errors of metabolism) concerns in primary care practice. Ann. Transl. Med. 2018, 6, 469. [Google Scholar] [CrossRef]
- Chakrapani, A.; Cleary, M.A.; Wraith, J.E. Detection of inborn errors of metabolism in the newborn. Arch. Dis. Child. Fetal Neonatal Ed. 2001, 84, F205–F210. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, S.; Matthijs, G.; Jaeken, J. Congenital disorders of glycosylation: A review. Pediatric Res. 2002, 52, 618–624. [Google Scholar] [CrossRef]
- Weinstein, D.A.; Steuerwald, U.; De Souza, C.F.M.; Derks, T.G.J. Inborn Errors of Metabolism with Hypoglycemia: Glycogen Storage Diseases and Inherit.ed Disorders of Gluconeogenesis. Pediatric Clin. N. Am. 2018, 65, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, L.P.; DeBrosse, S.D.; McCandless, S.E. Inborn Errors of Metabolism with Acidosis: Organic Acidemias and Defects of Pyruvate and Ketone Body Metabolism. Pediatric Clin. N. Am. 2018, 65, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Parmar, H.A.; Hoefling, N.; Srinivasan, A. Inborn errors of metabolism: Combining clinical and radiologic clues to solve the mystery. Am. J. Roentgenol. 2014, 203, W315–W327. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.T. Inborn errors presenting with liver dysfunction. Semin. Neonatol. 2002, 7, 49–63. [Google Scholar] [CrossRef]
- Mak, C.M.; Law, E.C.; Lee, H.H.; Siu, W.K.; Chow, K.M.; Au Yeung, S.K.; Ngan, H.Y.; Tse, N.K.; Kwong, N.S.; Chan, G.C.; et al. The first pilot study of expanded newborn screening for inborn errors of metabolism and survey of related knowledge and opinions of health care professionals in Hong Kong. Hong Kong Med. J. 2018, 24, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Romao, A.; Simon, P.E.A.; Góes, J.E.C.; Pinto, L.L.C.; Giugliani, R.; Luca, G.R.; Carvalho, F.L.C. Initial Clinical Presentation in Cases of Inborn Errors of Metabolism in a Reference Children’s Hospital: Still a Diagnostic Challenge. Rev. Paul. Pediatric 2017, 35, 258–264. [Google Scholar]
- Cleary, M.A.; Green, A. Developmental delay: When to suspect and how to investigate for an inborn error of metabolism. Arch. Dis. Child. 2005, 90, 1128–1132. [Google Scholar] [CrossRef]
- McDonald, L.; Rennie, A.; Tolmie, J.; Galloway, P.; McWilliam, R. Investigation of global developmental delay. Arch. Dis. Child. 2006, 91, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Van Karnebeek, C.D.; Shevell, M.; Zschocke, J.; Moeschler, J.B.; Stockler, S. The metabolic evaluation of the child with an intellectual developmental disorder: Diagnostic algorithm for identification of treatable causes and new digital resource. Mol. Genet. Metab. 2014, 111, 428–438. [Google Scholar] [CrossRef]
- Van Karnebeek, C.D.; Stockler, S. Treatable inborn errors of metabolism causing intellectual disability: A systematic literature review. Mol. Genet. Metab. 2012, 105, 368–381. [Google Scholar] [CrossRef]
- Yuan, Y.; Su, W.; Zhu, M. Threshold-free measures for assessing the performance of medical screening tests. Front. Public Health 2015, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; Marom, R.; Sutton, V.R. Newborn screening: A review of history, recent advancements, and future perspectives in the era of next generation sequencing. Curr. Opin. Pediatric 2016, 28, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Brosco, J.P.; Mattingly, M.; Sanders, L.M. Impact of specific medical interventions on reducing the prevalence of mental retardation. Arch. Pediatric Adolesc. Med. 2006, 160, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Rhead, W.J. Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: A global perspective. J. Inherit. Metab. Dis. 2006, 29, 370–377. [Google Scholar] [CrossRef]
- Mak, D.Y.; Sykes, J.; Stephenson, A.L.; Lands, L.C. The benefits of newborn screening for cystic fibrosis: The Canadian experience. J. Cyst. Fibros. 2016, 15, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.K.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014, 312, 729–738. [Google Scholar] [CrossRef]
- Pitt, J.J. Newborn screening. Clin. Biochem. Rev. 2010, 31, 57–68. [Google Scholar]
- Christopher, R.; Sankaran, B.P. An insight into the biochemistry of inborn errors of metabolism for a clinical neurologist. Ann. Indian Acad. Neurol. 2008, 11, 68–81. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant. Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C. Systems biology: Metab.onomics. Nature 2008, 455, 1054–1056. [Google Scholar] [CrossRef]
- Showalter, M.R.; Cajka, T.; Fiehn, O. Epimetabolites: Discovering metabolism beyond building and burning. Curr. Opin. Chem. Biol. 2017, 36, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; van Schaftingen, E.; Hanson, A.D. Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 2013, 9, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P.; Scalbert, A.; Herceg, Z. Measuring the exposome: A powerful basis for evaluating environmental exposures and cancer risk. Envion. Mol. Mutagen. 2013, 54, 480–499. [Google Scholar] [CrossRef]
- Li, B.; He, X.; Jia, W.; Li, H. Novel Applications of Metabolomics in Personalized Medicine: A Mini-Review. Molecules 2017, 22, 1173. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Malkawi, A.; Albast, N.; Al Bougha, S.; Lopata, A.; Dasouki, M.; Abdel Rahman, A.M. A targeted metabolomics approach for clinical diagnosis of inborn errors of metabolism. Anal. Chim Acta 2018, 1025, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metab.olic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef] [PubMed]
- Bulbul, S. Novel approach for Newborn Errors in Metabolism Screening (NEMS) by NMR: Clinical NEMS-by-NMR study in Turkey. Clin. Biochem. 2014, 47, 700–701. [Google Scholar] [CrossRef] [PubMed]
- Aygen, S.; Dürr, U.; Hegele, P.; Kunig, J.; Spraul, M.; Schäfer, H.; Krings, D.; Cannet, C.; Fang, F.; Schütz, B.; et al. NMR-Based Screening for Inborn Errors of Metabolism: Initial Results from a Study on Turkish Neonates. JIMD Rep. 2014, 16, 101–111. [Google Scholar] [Green Version]
- Allwood, J.W.; Goodacre, R. An introduction to liquid chromatography-mass spectrometry instrumentation applied in plant metabolomic analyses. Phytochem. Anal. 2010, 21, 33–47. [Google Scholar] [CrossRef]
- Gonzalez-Dominguez, R.; Sayago, A.; Fernandez-Recamales, A. Direct infusion mass spectrometry for metabolomic phenotyping of diseases. Bioanalysis 2017, 9, 131–148. [Google Scholar] [CrossRef]
- Begou, O.; Gika, H.G.; Wilson, I.D.; Theodoridis, G. Hyphenated MS-based targeted approaches in metabolomics. Analyst 2017, 142, 3079–3100. [Google Scholar] [CrossRef] [PubMed]
- Abdel Rahman, A.M.; Ryczko, M.; Pawling, J.; Dennis, J.W. Probing the hexosamine biosynthetic pathway in human tumor cells by multitargeted tandem mass spectrometry. ACS Chem. Biol. 2013, 8, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Abdel Rahman, A.M.; Pawling, J.; Ryczko, M.; Caudy, A.A.; Dennis, J.W. Targeted metabolomics in cultured cells and tissues by mass spectrometry: Method development and validation. Anal. Chim. Acta 2014, 845, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.D. Tandem mass spectroscopy in diagnosis and clinical research. Indian J. Clin. Biochem. 2015, 30, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Kortz, L.; Helmschrodt, C.; Ceglarek, U. Fast liquid chromatography combined with mass spectrometry for the analysis of metabolites and proteins in human body fluids. Anal. Bioanal. Chem. 2011, 399, 2635–2644. [Google Scholar] [CrossRef]
- Petrick, L.; Edmands, W.; Schiffman, C.; Grigoryan, H.; Perttula, K.; Yano, Y.; Dudoit, S.; Whitehead, T.; Metayer, C.; Rappaport, S. An untargeted metabolomics method for archived newborn dried blood spots in epidemiologic studies. Metabolomics 2017, 13, 27. [Google Scholar] [CrossRef]
- Annesley, T.; Diamandis, E.; Bachmann, L.; Hanash, S.; Hart, B.; Javahery, R.; Singh, R.; Smith, R. A Spectrum of Views on Clinical Mass Spectrometry. Clin. Chem. 2016, 62, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC-MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef]
- Nunes de Paiva, M.J.; Menezes, H.C.; Cardeal, Z.D. Sampling and analysis of metabolomes in biological fluids. Analyst 2014, 139, 3683–3694. [Google Scholar] [CrossRef] [Green Version]
- Ficicioglu, C. New tools and approaches to newborn screening: Ready to open Pandora’s box? Mol. Case Stud. 2017, 3, a001842. [Google Scholar] [CrossRef]
- Sharma, A.; Jaiswal, S.; Shukla, M.; Lal, J. Dried blood spots: Concepts, present status, and future perspectives in bioanalysis. Drug Test. Anal. 2014, 6, 399–414. [Google Scholar]
- Zakaria, R.; Allen, K.J.; Koplin, J.J.; Roche, P.; Greaves, R.F. Advantages and Challenges of Dried Blood Spot Analysis by Mass Spectrometry Across the Total Testing Process. EJIFCC 2016, 27, 288–317. [Google Scholar] [PubMed]
- Rogers, L.E.; Porter, F.S. Hereditary orotic aciduria. II. A urinary screening test. Pediatrics 1968, 42, 423–428. [Google Scholar] [PubMed]
- Chalmers, R.A.; Watts, R.W.; Lawson, A.M. A comprehensive screening method for detecting organic acidurias and other metabolic diseases in acutely sick infants and children. Ann. Clin. Biochem. 1977, 14, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Koulman, A.; Prentice, P.; Wong, M.C.Y.; Matthews, L.; Bond, N.J.; Eiden, M.; Griffin, J.L.; Dunger, D.B. The development and validation of a fast and robust dried blood spot based lipid profiling method to study infant metabolism. Metabolomics 2014, 10, 1018–1025. [Google Scholar] [CrossRef] [Green Version]
- Prentice, P.; Turner, C.; Wong, M.C.; Dalton, R.N. Stability of metabolites in dried blood spots stored at different temperatures over a 2-year period. Bioanalysis 2013, 5, 1507–1514. [Google Scholar] [CrossRef]
- Dénes, J.; Szabó, E.; Robinette, S.L.; Szatmári, I.; Szőnyi, L.; Kreuder, J.G.; Rauterberg, E.W.; Takáts, Z. Metabonomics of newborn screening dried blood spot samples: A novel approach in the screening and diagnostics of inborn errors of metabolism. Anal. Chem. 2012, 84, 10113–10120. [Google Scholar] [CrossRef]
- Oliveira, R.V.; Henion, J.; Wickremsinhe, E.R. Automated high-capacity on-line extraction and bioanalysis of dried blood spot samples using liquid chromatography/high-resolution accurate mass spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 2415–2426. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Hedenström, M.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Optimization of procedures for collecting and storing of CSF for studying the metabolome in ALS. Amyotroph. Lateral Scler. 2009, 10, 229–236. [Google Scholar] [CrossRef]
- Kawasaki, G.; Ichikawa, Y.; Yoshitomi, I.; Umeda, M. Metabolomics of Salivary Biomarkers in Yusho Patients. Fukuoka Igaku Zasshi 2015, 106, 144–148. [Google Scholar]
- Burlina, A.B.; Celato, A.; Polo, G.; Edini, C.; Burlina, A.P. The Utility of CSF for the Diagnosis of Primary and Secondary Monoamine Neurotransmitter Deficiencies. EJIFCC 2017, 28, 64–76. [Google Scholar] [PubMed]
- Nasheeda, C.M.; Philip, P.; Shenoy, R.D.; Shetty, S. Diagnostic Utility of Cord Blood Thyroid Stimulating Hormone in Congenital Hypothyroidism in the Era of Expanded Newborn Screening. Indian J. Clin. Biochem. 2018, 33, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Sahebekhtiari, N.; Nielsen, C.B.; Johannsen, M.; Palmfeldt, J. Untargeted Metabolomics Analysis Reveals a Link between ETHE1-Mediated Disruptive Redox State and Altered Metab.olic Regulation. J. Proteome Res. 2016, 15, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, M.J.; Lehotay, D.C.; Platt, B.A.; Fisher, L.; Tan, R.; Phillips, H.; Clarke, J.T. Ethylmalonic and methylsuccinic aciduria in ethylmalonic encephalopathy arise from abnormal isoleucine metabolism. Metabolism 1998, 47, 836–839. [Google Scholar] [CrossRef]
- Kennedy, A.D.; Miller, M.J.; Beebe, K.; Wulff, J.E.; Evans, A.M.; Miller, L.A.; Sutton, V.R.; Sun, Q.; Elsea, S.H. Metabolomic Profiling of Human Urine as a Screen for Multiple Inborn Errors of Metabolism. Genet. Test. Mol. Biomark. 2016, 20, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Gertsman, I.; Barshop, B.A. Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab. Dis. 2018, 41, 355–366. [Google Scholar] [CrossRef]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.C.D.G.; Keularts, I.M.L.W.; et al. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef]
- Hatam, N.; Shirvani, S.; Javanbakht, M.; Askarian, M.; Rastegar, M. Cost-utility analysis of neonatal screening program, shiraz university of medical sciences, shiraz, iran 2010. Iran. J. Pediatric 2013, 23, 493–500. [Google Scholar]
- Khneisser, I.; Adib, S.; Assaad, S.; Megarbane, A.; Karam, P. Cost-benefit analysis: Newborn screening for inborn errors of metabolism in Lebanon. J. Med. Screen. 2015, 22, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Cohan, N.; Karimi, M.; Khalili, A.H.; Falahzadeh, M.H.; Samadi, B.; Mahdavi, M.R. The efficacy of a neonatal screening programme in decreasing the hospitalization rate of patients with G6PD deficiency in southern Iran. J. Med. Screen. 2010, 17, 66–67. [Google Scholar] [CrossRef]
- Bentler, K.; Zhai, S.; Elsbecker, S.A.; Arnold, G.L.; Burton, B.K.; Vockley, J.; Cameron, C.A.; Hiner, S.J.; Edick, M.J.; Berry, S.A.; et al. 221 newborn-screened neonates with medium-chain acyl-coenzyme A dehydrogenase deficiency: Findings from the Inborn Errors of Metabolism Collaborative. Mol. Genet. Metab. 2016, 119, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcken, B.; Wiley, V. Fifty years of newborn screening. J. Paediatric Child. Health 2015, 51, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Knoll, D.; de Hora, M.; Kyle, C.; Glamuzina, E.; Webster, D. The Risk of Fatty Acid Oxidation Disorders and Organic Acidemias in Children with Normal Newborn Screening. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2017; Volume 35, pp. 53–58. [Google Scholar]
- Yoon, H.R.; Lee, K.R.; Kim, H.; Kang, S.; Ha, Y.; Lee, D.H. Tandem mass spectrometric analysis for disorders in amino, organic and fatty acid metabolism: Two year experience in South Korea. S. Asian J. Trop. Med. Public Health 2003, 34, 115–120. [Google Scholar]
- Cheng, K.H.; Liu, M.Y.; Kao, C.H.; Chen, Y.J.; Hsiao, K.J.; Liu, T.T.; Lin, H.Y.; Huang, C.H.; Chiang, C.C.; Ho, H.J.; et al. Newborn screening for methylmalonic aciduria by tandem mass spectrometry: 7 years’ experience from two centers in Taiwan. J. Chin. Med. Assoc. 2010, 73, 314–318. [Google Scholar] [CrossRef]
- Tarini, B.A.; Christakis, D.A.; Welch, H.G. State newborn screening in the tandem mass spectrometry era: More tests, more false positive results. Pediatrics 2006, 118, 448–456. [Google Scholar] [CrossRef]
- Tu, W.J.; He, J.; Chen, H.; Shi, X.D.; Li, Y. Psychological effects of false positive results in expanded newborn screening in China. PLoS ONE 2012, 7, e36235. [Google Scholar] [CrossRef]
- Lipstein, E.A.; Perrin, J.M.; Waisbren, S.E.; Prosser, L.A. Impact of false positive newborn metabolic screening results on early health care utilization. Genet. Med. 2009, 11, 716–721. [Google Scholar] [CrossRef]
- Mengreli, C.; Kanaka-Gantenbein, C.; Girginoudis, P.; Magiakou, M.A.; Christakopoulou, I.; Giannoulia-Karantana, A.; Chrousos, G.P.; Dacou-Voutetakis, C. Screening for congenital hypothyroidism: The significance of threshold limit in false-negative results. J. Clin. Endocrinol. Metab. 2010, 95, 4283–4290. [Google Scholar] [CrossRef]
- Shlomi, T.; Cabili, M.N.; Ruppin, E. Predicting metabolic biomarkers of human inborn errors of metabolism. Mol. Syst. Biol. 2009, 5, 263. [Google Scholar] [CrossRef]
- Fingerhut, R.; De Jesus Silva Arevalo, G.; Baumgartner, M.R.; Häberle, J.; Rohrbach, M.; Figueroa, A.W.; Fresse, E.M.; Polanco, O.L.; Torresani, T. Postprandial changes of amino acid and acylcarnitine concentrations in dried blood samples. J. Inherit. Metab. Dis. 2010, 33, S235–S239. [Google Scholar] [CrossRef]
- Thiboonboon, K.; Leelahavarong, P.; Wattanasirichaigoon, D.; Vatanavicharn, N.; Wasant, P.; Shotelersuk, V.; Pangkanon, S.; Kuptanon, C.; Chaisomchit, S.; Teerawattananon, Y.; et al. An Economic Evaluation of Neonatal Screening for Inborn Errors of Metabolism Using Tandem Mass Spectrometry in Thailand. PLoS ONE 2015, 10, e0134782. [Google Scholar] [CrossRef] [PubMed]
- Pandor, A.; Eastham, J.; Beverley, C.; Chilcott, J.; Paisley, S. Clinical effectiveness and cost-effectiveness of neonatal screening for inborn errors of metabolism using tandem mass spectrometry: A systematic review. Health Technol. Assess. 2004, 8, 1–121. [Google Scholar]
- Tiwana, S.K.; Rascati, K.L.; Park, H. Cost-effectiveness of expanded newborn screening in Texas. Value Health 2012, 15, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Newborn screening: Toward a uniform screening panel and system. Genet. Med. 2006, 8, 1S–252S.
- Do, K.T.; Kastenmüller, G.; Mook-Kanamori, D.O.; Yousri, N.A..; Theis, F.J.; Suhre, K.; Krumsiek, J. Network-based approach for analyzing intra- and interfluid metabolite associations in human blood, urine, and saliva. J. Proteome Res. 2015, 14, 1183–1194. [Google Scholar] [CrossRef]
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T.; et al. Metabolomics enables precision medicine: A White Paper, Community Perspective. Metabolomics 2016, 12, 149. [Google Scholar] [CrossRef]
- Boelens, J.J.; Orchard, P.J.; Wynn, R.F. Transplantation in inborn errors of metabolism: Current considerations and future perspectives. Br. J. Haematol. 2014, 167, 293–303. [Google Scholar] [CrossRef]
- Edmondson, C.; Davies, J.C. Current and future treatment options for cystic fibrosis lung disease: Latest evidence and clinical implications. Adv. Chronic Dis. 2016, 7, 170–183. [Google Scholar] [CrossRef]
- Ohashi, T. Gene therapy for lysosomal storage diseases and peroxisomal diseases. J. Hum. Genet. 2019, 64, 139–143. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Boutin, M. Novel gb(3) isoforms detected in urine of fabry disease patients: A metabolomic study. Curr. Med. Chem. 2012, 19, 3241–3252. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Boutin, M.; Gagnon, R.; Dupont, F.O.; Lavoie, P.; Clarke, J.T. Urinary globotriaosylsphingosine-related biomarkers for Fabry disease targeted by metabolomics. Anal. Chem. 2012, 84, 2745–2753. [Google Scholar] [CrossRef]
- Manwaring, V.; Boutin, M.; Auray-Blais, C. A metabolomic study to identify new globotriaosylceramide-related biomarkers in the plasma of Fabry disease patients. Anal. Chem. 2013, 85, 9039–9048. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Auray-Blais, C. Multiplex tandem mass spectrometry analysis of novel plasma lyso-Gb(3)-related analogues in Fabry disease. Anal. Chem. 2014, 86, 3476–3483. [Google Scholar] [CrossRef] [PubMed]
- Baig, F.; Pechlaner, R.; Mayr, M. Caveats of Untargeted Metabolomics for Biomarker Discovery. J. Am. Coll. Cardiol. 2016, 68, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Narath, S.H.; Mautner, S.I.; Svehlikova, E.; Schultes, B.; Pieber, T.R.; Sinner, F.M.; Gander, E.; Libiseller, G.; Schimek, M.G.; Sourij, H.; et al. An Untargeted Metabolomics Approach to Characterize Short-Term and Long-Term Metab.olic Changes after Bariatric Surgery. PLoS ONE 2016, 11, e0161425. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Alvarez, J.A.; Kang, J.; Yu, T. Network Marker Selection for Untargeted LC-MS Metabolomics Data. J. Proteome Res. 2017, 16, 1261–1269. [Google Scholar] [CrossRef]
- Andrisic, L.; Andrisic, L.; Dudzik, D.; Barbas, C.; Milkovic, L.; Grune, T.; Zarkovic, N. Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biol. 2018, 14, 47–58. [Google Scholar] [CrossRef]
- Korver-Keularts, I.M.l.W.; Wang, P.; Waterval, H.W.A.H.; Kluijtmans, L.A.J.; Wevers, R.A.; Langhans, C.D.; Scott, C.; Habets, D.D.J.; Bierau, J. Fast and accurate quantitative organic acid analysis with LC-QTOF/MS facilitates screening of patients for inborn errors of metabolism. J. Inherit. Metab. Dis. 2018, 41, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Keyfi, F.; Lukacs, Z.; Varasteh, A. A Description of Reference Ranges for Organic Acids in Urine Samples from A Pediatric Population in Iran. Rep. Biochem. Mol. Biol. 2017, 6, 40–50. [Google Scholar]
- Bachmann, C.; Buhlmann, R.; Colombo, J.P. Organic acids in urine: Sample preparation for GC/MS. J. Inherit. Metab. Dis. 1984, 7, 126. [Google Scholar] [PubMed]
- Wajner, M.; Sitta, A.; Kayser, A.; Deon, M.; Groehs, A.C.; Coelho, D.M.; Vargas, C.R. Screening for organic acidurias and aminoacidopathies in high-risk Brazilian patients: Eleven-year experience of a reference center. Genet. Mol. Biol. 2019, 42, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyniak, R.; Kosnowska, A.; Macioszek, S.; Bartoszewski, R.; Jan Markuszewski, M. New plasma preparation approach to enrich metabolome coverage in untargeted metabolomics: Plasma protein bound hydrophobic metabolite release with proteinase K. Sci. Rep. 2018, 8, 9541. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cruickshank, C.; Armstrong, M.; Mahaffey, S.; Reisdorph, R.; Reisdorph, N. New sample preparation approach for mass spectrometry-based profiling of plasma results in improved coverage of metabolome. J. Chromatogr. A 2013, 1300, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, S.; Vallejo, M.; García, A.; Barbas, C. Method validation strategies involved in non-targeted metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Marsal, S.; Julia, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, G.; Gika, H.G.; Wilson, I.D. Mass spectrometry-based holistic analytical approaches for metabolite profiling in systems biology studies. Mass Spectrom. Rev. 2011, 30, 884–906. [Google Scholar] [CrossRef]
- Rochat, B.; Mohamed, R.; Sottas, P.E. LC-HRMS Metabolomics for Untargeted Diagnostic Screening in Clinical Laboratories: A Feasibility Study. Metabolites 2018, 8, 39. [Google Scholar] [CrossRef]
- Barupal, D.K.; Fiehn, O. Chem.ical Similarity Enrichment Analysis (Chem.RICH) as alternative to biochemical pathway mapping for metabolomic datasets. Sci. Rep. 2017, 7, 14567. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Pirhaji, L.; Milani, P.; Leidl, M.; Curran, T.; Avila-Pacheco, J.; Clish, C.B.; White, F.M.; Saghatelian, A.; Fraenkel, E. Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat. Methods 2016, 13, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessandro, A.; Giardina, B.; Gevi, F.; Timperio, A.M.; Zolla, L. Clinical metabolomics: The next stage of clinical biochemistry. Blood Transfus. 2012, 10, S19–S24. [Google Scholar]
- Kim, S.J.; Kim, S.H.; Kim, J.H.; Hwang, S.; Yoo, H.J. Understanding Metabolomics in Biomedical Research. Endocrinol. Metab. 2016, 31, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.; Lee, J.; Price, M.; Tarailo-Graovac, M.; Matthews, A.; Engelke, U.; Tang, J.; Kluijtmans, L.A.J.; Wevers, R.A.; Wasserman, W.W.; et al. Integration of genomics and metabolomics for prioritization of rare disease variants: A 2018 literature review. J. Inherit. Metab. Dis. 2018, 41, 435–445. [Google Scholar] [CrossRef]
- Estrella, J.; Wilcken, B.; Carpenter, K.; Bhattacharya, K.; Tchan, M.; Wiley, V. Expanded newborn screening in New South. Wales: Missed Cases. J. Inherit. Metab. Dis. 2014, 37, 881–887. [Google Scholar] [CrossRef]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [Green Version]
- Janeckova, H.; Kalivodova, A.; Najdekr, L.; Friedecky, D.; Hron, K.; Bruheim, P.; Adam, T. Untargeted metabolomic analysis of urine samples in the diagnosis of some inherited metabolic disorders. Biomed. Pap. 2015, 159, 582–585. [Google Scholar] [CrossRef] [Green Version]
- Atwal, P.S.; Donti, T.R.; Cardon, A.L.; Bacino, C.A.; Sun, Q.; Emrick, L.; Reid Sutton, V.; Elsea, S.H. Aromatic L-amino acid decarboxylase deficiency diagnosed by clinical metabolomic profiling of plasma. Mol. Genet. Metab. 2015, 115, 91–94. [Google Scholar] [CrossRef]
- Najdekr, L.; Gardlo, A.; Mádrová, L.; Friedecký, D.; Janečková, H.; Correa, E.S.; Goodacre, R.; Adam, T. Oxidized phosphatidylcholines suggest oxidative stress in patients with medium-chain acyl-CoA dehydrogenase deficiency. Talanta 2015, 139, 62–66. [Google Scholar] [CrossRef]
- Donti, T.R.; Cappuccio, G.; Hubert, L.; Neira, J.; Atwal, P.S.; Miller, M.J.; Cardon, A.L.; Sutton, V.R.; Porter, B.E.; Baumer, F.M.; et al. Diagnosis of adenylosuccinate lyase deficiency by metabolomic profiling in plasma reveals a phenotypic spectrum. Mol. Genet. Metab. Rep. 2016, 8, 61–66. [Google Scholar] [CrossRef]
- McCoin, C.S.; Piccolo, B.D.; Knotts, T.A.; Matern, D.; Vockley, J.; Gillingham, M.B.; Adams, S.H. Unique plasma metabolomic signatures of individuals with inherited disorders of long-chain fatty acid oxidation. J. Inherit. Metab. Dis. 2016, 39, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappuccio, G.; Pinelli, M.; Alagia, M.; Donti, T.; Day-Salvatore, D.L.; Veggiotti, P.; De Giorgis, V.; Lunghi, S.; Vari, M.S.; Striano, P.; et al. Biochemical phenotyping unravels novel metabolic abnormalities and potential biomarkers associated with treatment of GLUT1 deficiency with ketogenic diet. PLoS ONE 2017, 12, e0184022. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Abily-Donval, L.; Schmitz-Afonso, I.; Héron, B.; Piraud, M.; Ausseil, J.; Zerimech, F.; Gonzalez, B.; Marret, S.; Afonso, C.; et al. Unveiling metabolic remodeling in mucopolysaccharidosis type III through integrative metabolomics and pathway analysis. J. Transl. Med. 2018, 16, 248. [Google Scholar] [CrossRef] [PubMed]
- Burrage, L.C.; Thistlethwaite, L.; Stroup, B.M.; Sun, Q.; Miller, M.J.; Nagamani, S.C.S.; Craigen, W.; Scaglia, F.; Sutton, V.R.; Graham, B.; et al. Untargeted metabolomic profiling reveals multiple pathway perturbations and new clinical biomarkers in urea cycle disorders. Genet. Med. 2019, 21, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Vaclavik, J.; Coene, K.L.M.; Vrobel, I.; Najdekr, L.; Friedecký, D.; Karlíková, R.; Mádrová, L.; Petsalo, A.; Engelke, U.F.H.; van Wegberg, A.; et al. Structural elucidation of novel biomarkers of known metabolic disorders based on multistage fragmentation mass spectra. J. Inherit. Metab. Dis. 2018, 41, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Sandlers, Y. The future perspective: Metabolomics in laboratory medicine for inborn errors of metabolism. Transl. Res. 2017, 189, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Ogundare, M.; Williams, C.M.; Wang, Y. On the future of “omics”: Lipidomics. J. Inherit. Metab. Dis. 2011, 34, 583–592. [Google Scholar] [CrossRef]
- Lamari, F.; Mochel, F.; Saudubray, J.M. An overview of inborn errors of complex lipid biosynthesis and remodelling. J. Inherit. Metab. Dis. 2015, 38, 3–18. [Google Scholar] [CrossRef]
- Herzog, K.; Pras-Raves, M.L.; Ferdinandusse, S.; Vervaart, M.A.T.; Luyf, A.C.M.; van Kampen, A.H.C.; Wanders, R.J.A.; Waterham, H.R.; Vaz, F.M. Plasma lipidomics as a diagnostic tool for peroxisomal disorders. J. Inherit. Metab. Dis. 2018, 41, 489–498. [Google Scholar] [CrossRef]
- Lydic, T.A.; Goo, Y.H. Lipidomics unveils the complexity of the lipidome in metabolic diseases. Clin. Transl. Med. 2018, 7, 4. [Google Scholar] [CrossRef]
- Li, M.; Yang, L.; Bai, Y.; Liu, H. Analytical methods in lipidomics and their applications. Anal. Chem. 2014, 86, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Rashed, M.S.; Ozand, P.T.; Bennett, M.J.; Barnard, J.J.; Govindaraju, D.R.; Rinaldo, P. Inborn errors of metabolism diagnosed in sudden death cases by acylcarnitine analysis of postmortem bile. Clin. Chem. 1995, 41, 1109–1114. [Google Scholar] [PubMed]
- Byeon, S.K.; Kim, J.Y.; Lee, J.S.; Moon, M.H. Variations in plasma and urinary lipids in response to enzyme replacement therapy for Fabry disease patients by nanoflow UPLC-ESI-MS/MS. Anal. Bioanal. Chem. 2016, 408, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Seyer, A.; Boudah, S.; Broudin, S.; Junot, C.; Colsch, B. Annotation of the human cerebrospinal fluid lipidome using high resolution mass Spectrom.etry and a dedicated data processing workflow. Metabolomics 2016, 12, 91. [Google Scholar] [CrossRef]
- Mandal, R.; Chamot, D.; Wishart, D.S. The role of the Human Metab.olome Database in inborn errors of metabolism. J. Inherit. Metab. Dis. 2018, 41, 329–336. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef]
- Pluskal, T.; Castilol, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass Spectrom.etry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Clasquin, M.F.; Melamud, E.; Rabinowitz, J.D. LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinformatics 2012, 37. [Google Scholar] [CrossRef]
- Dias, D.A.; Jones, O.A.H.; Beale, D.L.; Boughton, B.A.; Benheim, D.; Kouremenos, K.A.; Wolfender, J.-L.; Wishart, D.S. Current and Future Perspectives on the Structural Identification of Small Molecules in Biological Systems. Metabolites 2016, 6, 46. [Google Scholar] [CrossRef]
- Blazenovic, I.; Kind, T.; Sa, M.R.; Ji, J.; Vaniya, A.; Wancewicz, B.; Roberts, B.S.; Torbašinović, H.; Lee, T.; Mehta, S.S.; et al. Structure Annotation of All Mass Spectra in Untargeted Metabolomics. Anal. Chem. 2019, 91, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Liu, K.H.; Lee, D.P.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass Spectrom.etry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metab.olome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metab.olome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2018, 37, 513–532. [Google Scholar] [CrossRef]
- Smith, C.A.; Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Drug Monit 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Jeffryes, J.G.; Colastani, R.L.; Elbadawi-Sidhu, M.; Kind, T.; Niehaus, T.D.; Broadbelt, L.J.; Hanson, A.D.; Fiehn, O.; Tyo, K.E.; Henry, C.S. MINEs: Open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015, 7, 44. [Google Scholar] [CrossRef]
- Huan, T.; Tang, C.; Li, R.; Shi, Y.; Lin, G.; Li, L. MyCompoundID MS/MS Search: Metabolite Identification Using a Library of Predicted Fragment-Ion.-Spectra of 383,830 Possible Human Metabolites. Anal. Chem. 2015, 87, 10619–10626. [Google Scholar] [CrossRef]
- Aretz, I.; Meierhofer, D. Advantages and Pitfalls of Mass Spectrom.etry Based Metab.olome Profiling in Systems Biol.ogy. Int. J. Mol. Sci. 2016, 17, 632. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.; Wang, C.; Zhang, H.; Cai, Z. Non-targeted and targeted metabolomics approaches to diagnosing lung cancer and predicting patient prognosis. Oncotarget 2016, 7, 63437–63448. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Riano, C.; Sanz-Rodríguez, M.; Escudero-Ramirez, J.; Lorenzo, M.P.; Barbas, C.; Cubelos, B.; Garcia, A. Target. and untargeted GC-MS based metabolomic study of mouse optic nerve and its potential in the study of neurological visual diseases. J. Pharm. Biomed. Anal. 2018, 153, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.; Farrell, P.M. The magnitude and challenge of false positive newborn screening test results. Arch. Pediatric Adolesc. Med. 2000, 154, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.L.; Castellanos-Brown, K.; Childress, S.; Bonhomme, N.; Oktay, J.S.; Terry, S.F.; Kyler, P.; Davidoff, A.; Greene, C. The impact of false positive newborn screening results on families: A qualitative study. Genet. Med. 2012, 14, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Rock, M.J.; Levy, H.; Zaleski, C.; Farrell, P.M. Factors accounting for a missed diagnosis of cystic fibrosis after newborn screening. Pediatric Pulmonol. 2011, 46, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.D.; Pappan, K.L.; Donti, T.R.; Evans, A.M.; Wulff, J.E.; Miller, L.A.D.; Reid Sutton, V.; Sun, Q.; Miller, M.J.; Elsea, S.H. Elucidation of the complex metabolic profile of cerebrospinal fluid using an untargeted biochemical profiling assay. Mol. Genet. Metab. 2017, 121, 83–90. [Google Scholar] [CrossRef]
- Percenti, L.; Vickery, G. Newborn Screening Follow-up. N. C. Med. J. 2019, 80, 37–41. [Google Scholar] [CrossRef]
- Pappan, K.L.; Kennedy, A.D.; Magoulas, P.L.; Hanchard, N.A.; Sun, Q.; Elsea, S.H. Clinical Metabolomics to Segregate Aromatic Amino Acid Decarboxylase Deficiency From Drug-Induced Metabolite Elevations. Pediatric Neurol. 2017, 75, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Sweetman, L. Newborn screening by tandem mass Spectrom.etry: Gaining experience. Clin. Chem. 2001, 47, 1937–1938. [Google Scholar]
- Dupont, F.O.; Gagnon, R.; Boutin, M.; Auray-Blais, C. A metabolomic study reveals novel plasma lyso-Gb3 analogs as Fabry disease biomarkers. Curr. Med. Chem. 2013, 20, 280–288. [Google Scholar] [CrossRef]
- Lavoie, P.; Boutin, M.; Auray-Blais, C. Multiplex analysis of novel urinary lyso-Gb3-related biomarkers for Fabry disease by tandem mass Spectrom.etry. Anal. Chem. 2013, 85, 1743–1752. [Google Scholar] [CrossRef]
- Li, H.; Zhao, L.; Singh, R.; Ham, J.N.; Fadoju, D.O.; Bean, L.J.H.; Zhang, Y.; Xu, Y.; Xu, H.E.; Gambello, M.J. The first pediatric case of glucagon receptor defect due to biallelic mutations in GCGR is identified by newborn screening of elevated arginine. Mol. Genet. Metab. Rep. 2018, 17, 46–52. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
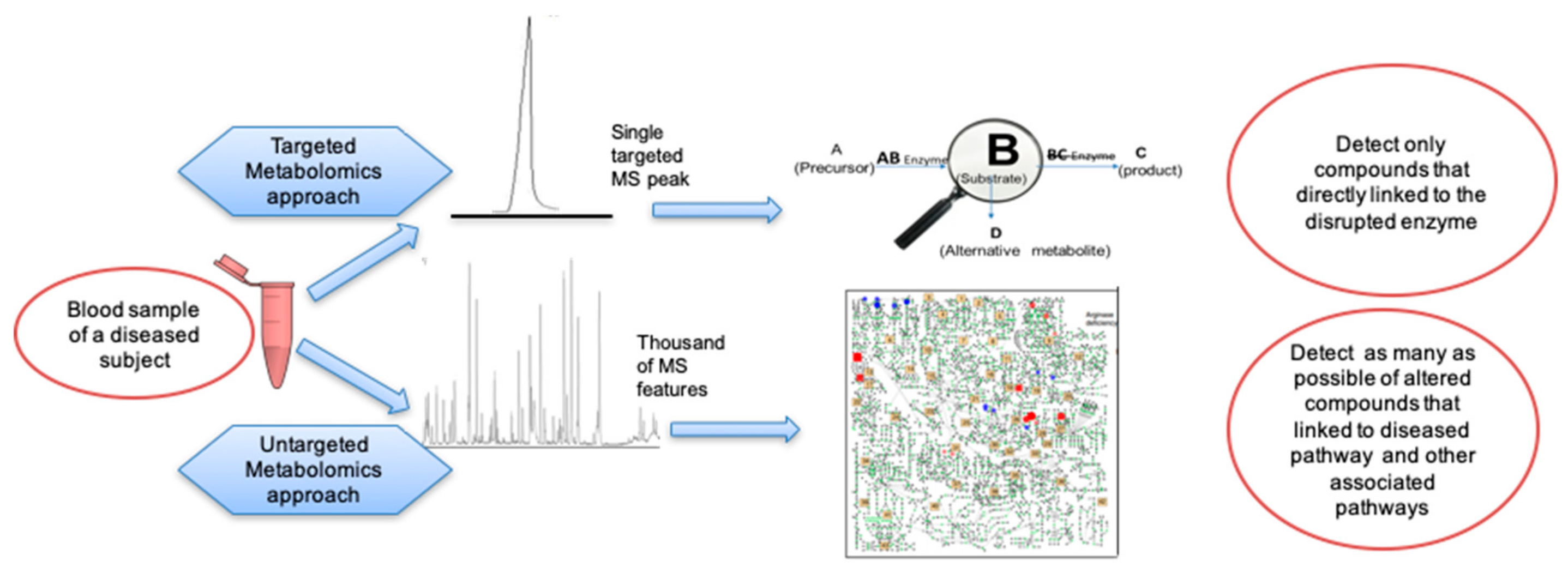
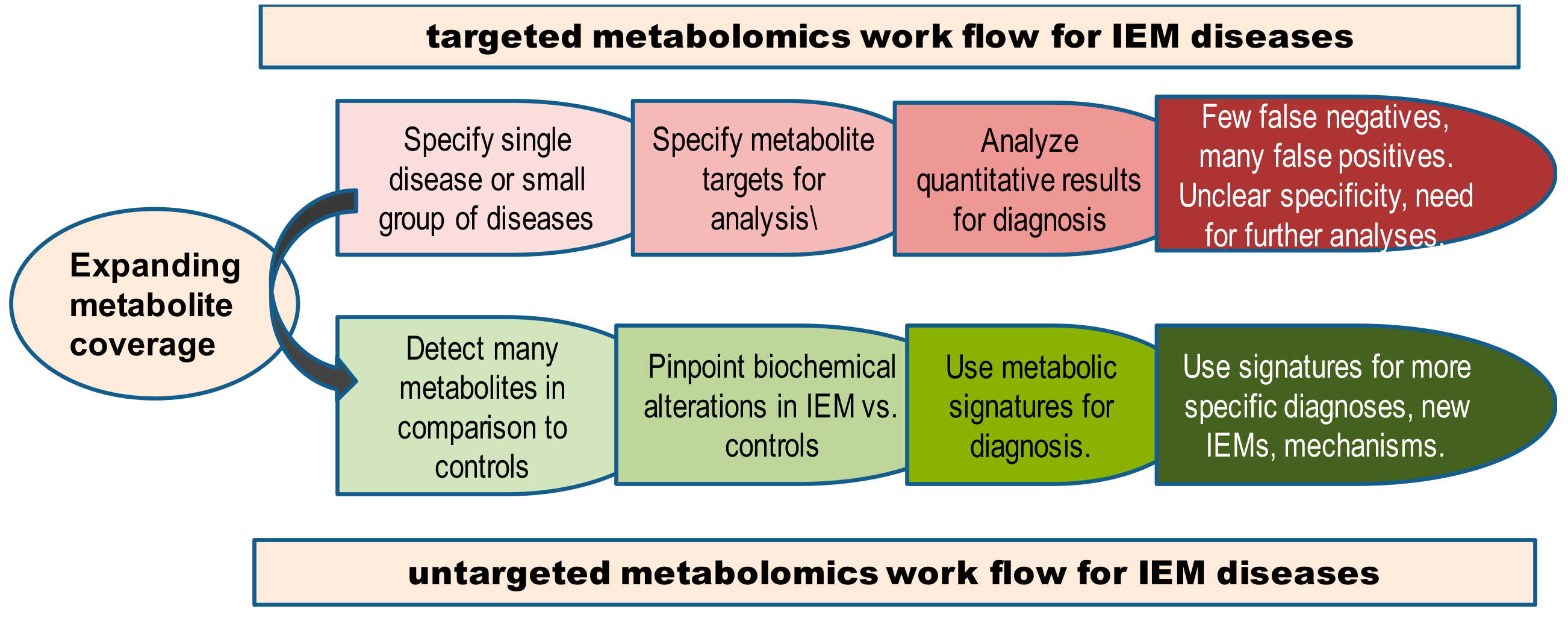
| Parameter | Targeted Metabolomics | Untargeted Metabolomics |
|---|---|---|
| Main concept | Select specific metabolites (10-100) as targets in LC-MS/MS or direct infusion MS/MS to diagnose a specific disease. Detect fragment ions of these metabolic targets and perform molar quantification using internal standards | Detect all ions within a certain mass range in LC-MS/MS and identify as many metabolites as possible. Use signal intensities of both known and unknown metabolites to characterize diseases phenotypes. Quantification can be aided by quality controls, normalizations, and internal standards. |
| Instrumentation | GC-MS (in single ion monitoring) LC-triple quadrupole MS LC-quadrupole linear ion trap MS (in multi-reaction monitoring) | GC-MS (full scan) LC-quadrupole time-of-flight MS LC-orbital ion trap MS |
| Weaknesses | Selective isolation of a group of metabolites. Focus on only specific (target) metabolites may increase the risk of overlooking metabolic responses in other pathways. Target metabolites may lack specificity to classify a variety of IEMs. Costs for internal standards and complexity of data analysis increases with the number of target metabolites. | Maximum number of metabolites. Relative (normalized) signal intensities are not robust inter-laboratory units. Lack of absolute quantification hampers defining ‘normal’ metabolite levels on a population level. Comparisons only based on differentiating groups within studies. Data processing parameters not validated across different software. Compound identification is not standardized yet. |
| Strengths | Hypothesis testing: Targeted experiments provide better quantitation, typically by internal standards and specific mass spectrometer conditions. Absolute quantifications of metabolite in may be used to establish baseline metabolite levels for defining healthy versus altered states and for interlaboratory comparison. Identification is performed by comparison to internal standards and specificity of MS/MS. | Hypothesis generating: Untargeted experiments provide broader coverage with the potential to screen known compounds and discover novel metabolites. Cover “all” metabolites in samples within the bounds of an analytical technique. Typically >1000 metabolite signals. No increase in the cost when more metabolites are detected. More information about the overall genomic environmental interaction to yield specific IEM phenotypes. |
| Sample | Instrumentation and Platform | Number of Samples | Number of Studied Diseases | Results | ref. |
|---|---|---|---|---|---|
| Plasma | LC ESI (−) QTOF C18 column, | 24 patients, 21 controls | 9 patients with propionic academia, 15 patients with methylmalonic acidemia | Classification by known and new markers | [42] |
| Dried blood spots | ESI (+,−) Orbitrap Q-Exactive MS | 66 patients, 500 controls | 9 diseases: PKU, MCADD, HCY, CLD, MSUD, IVA, PA, 3-MCC, Tyrosinemia, citrullinemia galactosemia | Correctly grouped previous false positive cases | [108] |
| Urine | LC ESI (+) QTOF HILIC amide column | 21 patients, 14 controls | 4 diseases: cystinuria, maple syrup urine disease, adenylosuccinate lyase deficiency, galactosemia | Groups were correctly classified | [169] |
| Plasma | GC-MS, ESI (+,−) Orbitrap MS HILIC column | 1 patient | Aromatic L-amino acid decarboxylase (AADC) deficiency | Case study | [170] |
| Plasma | GC-MS, ESI (+,−) LC-MS HILIC column | 120 patients 70 controls | 21 IEM diseases | 20 IEMs classified, novel biomarkers | [168] |
| Dried blood spots | ESI (+) Orbitrap MS | 25 patients 25 controls | Medium Chain Acyl-COA Dehydrogenase Deficiency (MCADD) | Disease groups classified | [171] |
| Plasma | GC-MS, ESI (+,−) Orbitrap MS HILIC column | 4 patients | Adenyl succinate lyase (ADSL) deficiency | Disease characterized | [172] |
| Plasma | GC-MS, lipidomics by LC-QTOF MS | 12 patients, 11 controls | Long-Chain Hydroxy Acyl CoA Dehydrogenase, Carnitine Palmitoyl Transferase 2 Deficiency | Identified with pathway detection | [173] |
| Urine | LC ESI (+,−) Q-Exactive MS HILIC column | 34 patients 66 controls | 18 IEM diseases | Characterization | [116] |
| Skin fibroblasts | LC-ESI (+,−) QTOF MS with HILIC column | 3 patients 3 controls | Ethylmalonic Encephalopathy | Detected possible new biomarker | [114] |
| CSF, urine plasma | GC-MS, LC (+,−) ESI Orbitrap w/ HILIC column | 17 patients | Glucose Transporter Type 1 Deficiency Syndrome (GLUT1-DS) | Detected possible new biomarker, pathway affected | [174] |
| Urine | LC ion mobility MS | 49 patients 66 controls | Mucopolysaccharidosis MPS III A, B, C, D | Four phenotypes identified with pathways | [175] |
| Plasma | LC (+,−) QTOF HILIC column | 46 IEM diseases | 42 IEM groups, new biomarkers | [118] | |
| Plasma | LC - heated ESI Q-Exactive MS | 48 patients | Various types of urea cycle defect (UCD) | Detect novel metabolites, monitor treatment | [176] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, I.T.; Showalter, M.R.; Fiehn, O. Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites 2019, 9, 242. https://doi.org/10.3390/metabo9100242
Ismail IT, Showalter MR, Fiehn O. Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites. 2019; 9(10):242. https://doi.org/10.3390/metabo9100242
Chicago/Turabian StyleIsmail, Israa T, Megan R Showalter, and Oliver Fiehn. 2019. "Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics" Metabolites 9, no. 10: 242. https://doi.org/10.3390/metabo9100242
APA StyleIsmail, I. T., Showalter, M. R., & Fiehn, O. (2019). Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites, 9(10), 242. https://doi.org/10.3390/metabo9100242