Advances of Metabolomics in Fungal Pathogen–Plant Interactions



Abstract
:1. Introduction
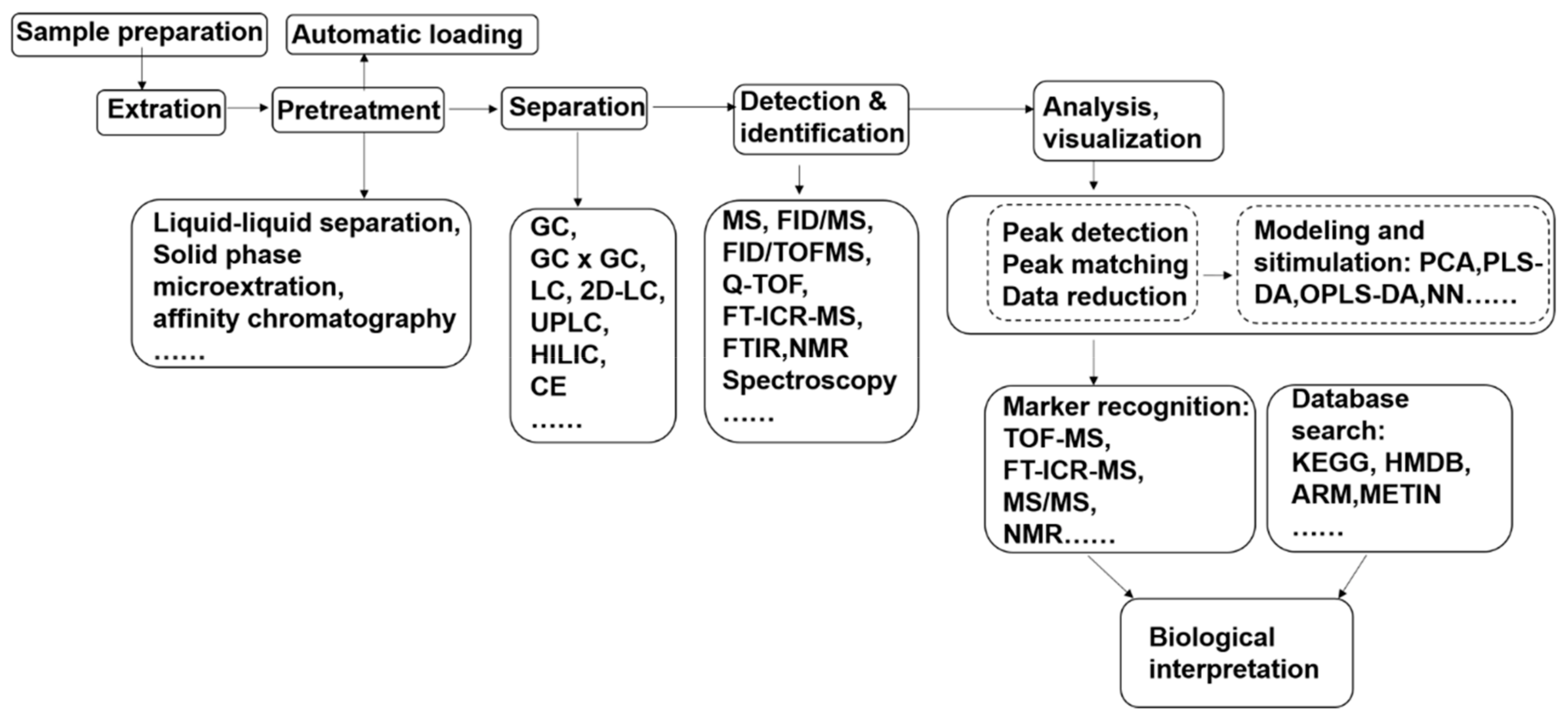
2. Metabolomics Methods for Fungal Pathogen–Plant Interactions
2.1. Experimental Design
2.2. Sample Preparation
2.3. Data Collection
2.4. Data Processing and Analysis
3. Research Progress and Application of Metabolomics in Fungal Pathogen–Plant Interactions
3.1. Progress in Metabolomics Research for Fungal Pathogen–Plant Interactions
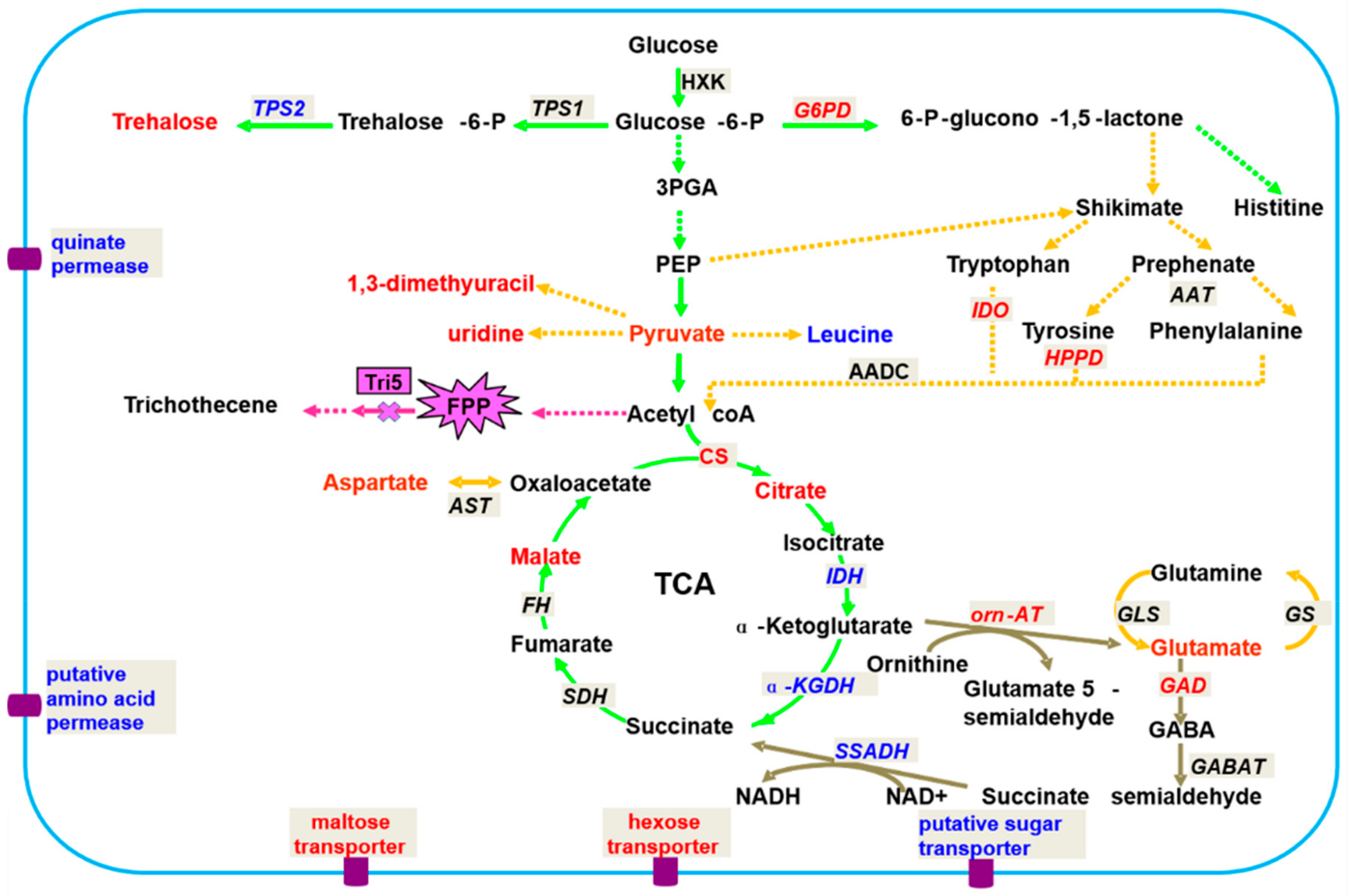
3.1.1. Fusarium graminearum–Wheat Interaction
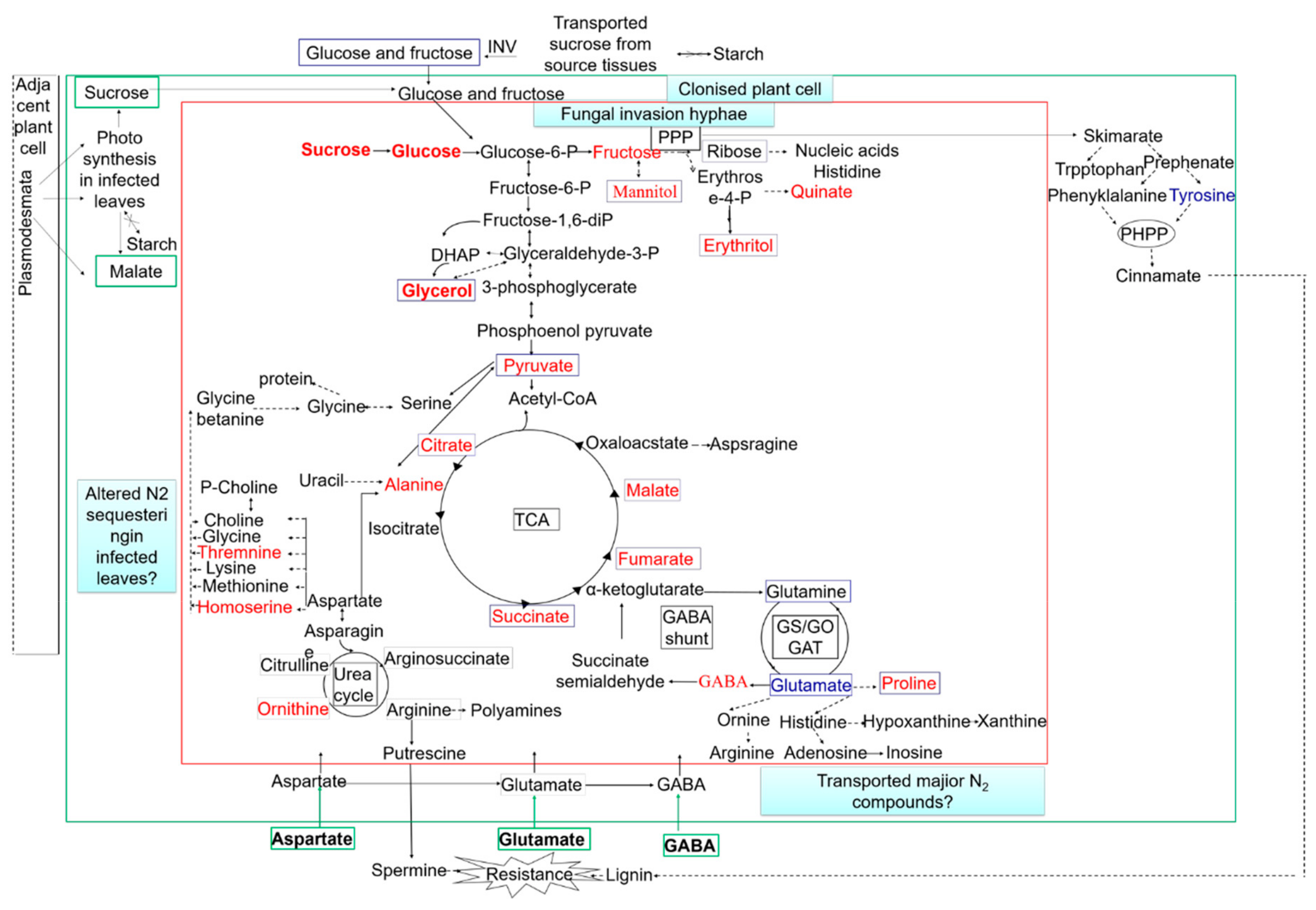
3.1.2. Magnaporthe oryzae–Rice Interaction
3.1.3. Ustilago maydis–Maize Interaction
3.1.4. Rhizoctonia solani–Plant Interaction
3.1.5. Botrytis cinerea–Plant Interaction
3.1.6. Other Fungal Pathogen–Plant Interactions
3.1.7. Integrating Multi-Omics Assisted Metabolomics Research of Fungal Pathogen–Plant Interactions
4. Prospects and Challenges
Author Contributions
Funding
Conflicts of Interest
References
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genom. 2001, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, A.L.; Robison, F.M.; Lyons, S.M.; Broeckling, C.D.; Prenni, J.E. Evaluating plant immunity using mass spectrometry-based metabolomics workflows. Front. Plant Sci. 2014, 5, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baidoo, E.E.K. Microbial Metabolomics: A General Overview. Methods Mol. Biol. 2019, 1859, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Merlo, M.E.; Jankevics, A.; Takano, E.; Breitling, R. Exploring the metabolic state of microorganisms using metabolomics. Bioanalysis 2011, 3, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.F.; Larsen, T.O. The importance of mass spectrometric dereplication in fungal secondary metabolite analysis. Front. Microbiol. 2015, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Yi, P.S.; Tang, H.; Wang, Y. Recent developments in sample preparation and data pre-treatment in metabonomics research. Arch. Biochem. Biophys. 2016, 589, 4–9. [Google Scholar] [CrossRef]
- Roessner, U.; Wagner, C.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Technical advance: Simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 2010, 23, 131–142. [Google Scholar] [CrossRef]
- Werf, M.J.V.D.; Jellema, R.H.; Hankemeier, T. Microbial metabolomics: Replacing trial-and-error by the unbiased selection and ranking of targets. J. Ind. Microbiol. Biotechnol. 2005, 32, 234–252. [Google Scholar] [CrossRef]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.F.; Zhang, J.T.; Song, X.S.; Yang, J.; Li, H.P.; Tang, H.R.; Liao, Y.C. Combined metabonomic and quantitative real-time PCR analyses reveal systems metabolic changes of Fusarium graminearum induced by tri5 gene deletion. J. Proteome Res. 2011, 10, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
- Kuhara, T.; Ohse, M.; Inoue, Y.; Cooper, A.J. A GC/MS-based metabolomic approach for diagnosing citrin deficiency. Anal. Bioanal. Chem. 2011, 400, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Halket, J.M.; Waterman, D.; Przyborowska, A.M.; Patel, R.K.; Fraser, P.D.; Bramley, P.M. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J. Exp. Bot. 2005, 56, 219. [Google Scholar] [CrossRef] [PubMed]
- Simo, C.; Ibanez, C.; Gomez-Martinez, A.; Ferragut, J.A.; Cifuentes, A. Is metabolomics reachable? Different purification strategies of human colon cancer cells provide different ce-ms metabolite profiles. Electrophoresis 2011, 32, 1765–1777. [Google Scholar] [CrossRef] [PubMed]
- Perrett, D.; Ross, G. Capillary electrophoresis: A powerful tool for biomedical analysis and research? TrAC Trends Anal. Chem. 1992, 11, 156–163. [Google Scholar] [CrossRef]
- Johnson, H.E.; Broadhurst, D.; Goodacre, R.; Smith, A.R. Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 2003, 62, 919–928. [Google Scholar] [CrossRef]
- Water, N.J.; Holmes, E.; Williams, A.; Waterfield, N.J.; Farrant, R.D.; Nicholson, J.K. NMR and pattern recognition studies on the time-related metabolic effects of α-Naphthylisothiocyanate on liver, urine, and plasma in the rat: An integrative metabonomic approach. Chem. Res. Toxicol. 2001, 14, 1401–1412. [Google Scholar] [CrossRef]
- Goodacre, R. Metabolomics of a Superorganism. J. Nutr. 2007, 137, 259S. [Google Scholar] [CrossRef]
- Arjen, L. Metalign: Interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Anal. Chem. 2009, 81, 3079–3086. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. Mzmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Hall, R. Plant metabolomics: The missing link in functional genomics strategies. Plant Cell 2002, 14, 1437–1440. [Google Scholar] [CrossRef]
- Ralf, T.; Patti, G.J.; Duane, R.; Gary, S. XCMS online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef]
- Wei, X.L.; Shi, X.; Kim, S.; Zhang, L.; Patrick, J.S.; Binkley, J.; McClain, C.; Zhang, X. Data preprocessing method for liquid chromatography-mass spectrometry based metabolomics. Anal. Chem. 2012, 84, 7963–7971. [Google Scholar] [CrossRef] [PubMed]
- Broeckling, C.D.; Reddy, I.R.; Duran, A.L.; Xuechun, Z.; Sumner, L.W. Met-idea: Data extraction tool for mass spectrometry-based metabolomics. Anal. Chem. 2006, 78, 4334–4341. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.L.; Jian, Y.; Wang, L.; Sumner, L.W. Metabolomics spectral formatting, alignment and conversion tools (MSFACTs). Bioinformatics 2003, 19, 2283–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Dixon, R.A.; Willmitzer, L. Plant metabolomics volume 57 || gas chromatography mass spectrometry. Biotechnol. Agric. For. 2006, 57, 3–20. [Google Scholar] [CrossRef]
- Want, E.J.; Nordström, A.; Morita, H.; Siuzdak, G. From exogenous to endogenous: The inevitable imprint of mass spectrometry in metabolomics. J. Proteome Res. 2007, 6, 459–468. [Google Scholar] [CrossRef]
- Mastrangelo, A.; Ferrarini, A.; Rey-Stolle, F.; García, A.; Barbas, C. From sample treatment to biomarker discovery: A tutorial for untargeted metabolomics based on GC-(EI)-Q-MS. Anal. Chim. Acta 2015, 900, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; King, R.D.; Altmann, T.; Fiehn, O. Application of metabolomics to plant genotype discrimination using statistics and machine learning. Bioinformatics 2002, 18 (Suppl. S2), S241–S248. [Google Scholar] [CrossRef]
- Fukusaki, E.; Kobayashi, A. Plant metabolomics: Potential for practical operation. J. Biosci. Bioeng. 2005, 100, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.X.; An, Y.P.; Li, N.; Liu, B.F.; Wang, Y.L.; Tang, H.R. Multiple univariate data analysis reveals the inulin effects on the high-fat-diet induced metabolic alterations in rat myocardium and testicles in the preobesity state. J. Proteome R. 2013, 12, 3480–3495. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.M.; Ram, K.; Wishart, D.S. ECMDB: The E. coli metabolome database. Nucleic Acids Res. 2013, 41, D625–D630. [Google Scholar] [CrossRef] [PubMed]
- Shifrin, V.I.; Anderson, P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem. 1999, 274, 13985–13992. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Q.; Li, Y.W.; Luo, X.Y.; Yoshizawa, T. Fusarium toxins in wheat from an area in henan province, pr china, with a previous human red mould intoxication episode. Food Addit. Contam. 2002, 19, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Song, X.S.; Xing, S.; Li, H.P.; Zhang, J.B.; Qu, B.; Jiang, J.H.; Fan, C.; Yang, P.; Liu, J.L.; Hu, Z.Q. An antibody that confers plant disease resistance targets a membrane-bound glyoxal oxidase in fusarium. New Phytol. 2016, 210, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Wang, Z.; Liu, C. Roles of peroxisomes in the rice blast fungus. BioMed Res. Int. 2016, 2016, 9343417. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, R.; Saito, K. Integrated metabolomics for abiotic stress responses in plants. Curr. Opin. Plant Biol. 2015, 24, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feussner, I.; Polle, A. What the transcriptome does not tell - proteomics and metabolomics are closer to the plants’ patho-phenotype. Curr. Opin. Plant Biol. 2015, 26, 26–31. [Google Scholar] [CrossRef]
- Tan, K.C.; Ipcho, S.V.; Trengove, R.D.; Oliver, R.P.; Solomon, P.S. Assessing the impact of transcriptomics, proteomics and metabolomics on fungal phytopathology. Mol. Plant Pathol. 2010, 10, 703–715. [Google Scholar] [CrossRef]
- Smith, L.L. Key challenges for toxicologists in the 21st century. Trends Pharmacol. Sci. 2001, 22, 281–285. [Google Scholar] [CrossRef]
- Tang, H.R.; Wang, Y.L. Metabonomics: A Revolution in Progress. Prog. Biochem. Biophys. 2006, 33, 401–417. [Google Scholar] [CrossRef]
- David, P.; Manfred, B.; Enot, D.P.; Overy, D.P.; Zaira Caracuel, R.; Martin, G.; Nicholas, T.; John, D. Rice blast infection of Brachypodium distachyon as a model system to study dynamic host/pathogen interactions. Nat. Protoc. 2008, 3, 435–445. [Google Scholar] [CrossRef]
- Chen, F.F.; Liu, X.; Zhang, J.T.; FLei, H.H.; Li, H.P.; Tang, H.R.; Liao, Y.C. Combined metabonomic and quantitative rt-pcr analyses revealed metabolic reprogramming associated with Fusarium graminearum resistance in transgenic Arabidopsis thaliana. Front. Plant Sci. 2018, 8, 2177. [Google Scholar] [CrossRef] [PubMed]
- Allwood, J.W.; Ellis, D.I.; Goodacre, R. Metabolomic technologies and their application to the study of plants and plant-host interactions. Physiol. Plantarum. 2010, 132, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Shaner, G. Management and resistance in wheat and barley to fusarium head blight. Annu. Rev. Phytopathol. 2004, 42, 135–161. [Google Scholar] [CrossRef]
- Lowe, R.G.T.; Allwood, J.W.; Galster, A.M.; Urban, M.; Daudi, A.; Canning, G.; Ward, J.L.; Beale, M.H.; Hammondkosack, K.E. A combined 1h nuclear magnetic resonance and electrospray ionization-mass spectrometry analysis to understand the basal metabolism of plant-pathogenic fusarium spp. Mol. Plant Microbe Interact. 2010, 23, 1605–1618. [Google Scholar] [CrossRef]
- Balmer, D.; Flors, V.; Glauser, G.; Mauch-Mani, B. Metabolomics of cereals under biotic stress: Current knowledge and techniques. Front. Plant Sci. 2013, 4, 82. [Google Scholar] [CrossRef]
- Buerstmayr, M.; Lemmens, M.; Steiner, B.; Buerstmayr, H. Advanced backcross qtl mapping of resistance to fusarium head blight and plant morphological traits in a Triticum macha. Theor. Appl. Genet. 2011, 123, 293–306. [Google Scholar] [CrossRef]
- Li, X.; Luo, H.; Huang, T.; Xu, L.; Shi, X.; Hu, K. Statistically correlating NMR spectra and LC-MS data to facilitate the identification of individual metabolites in metabolomics mixtures. Anal. Bioanal. Chem. 2019, 411, 1301–1309. [Google Scholar] [CrossRef]
- Li, J.; Duan, Y.; Bian, C.; Pan, X.; Yao, C.; Wang, J.; Zhou, M. Effects of validamycin in controlling fusarium head blight caused by Fusarium graminearum: Inhibition of DON biosynthesis and induction of host resistance. Pestic. Biochem. Physiol. 2019, 153, 9. [Google Scholar] [CrossRef] [PubMed]
- Tomas, C.; Marta, V.; Zbynek, D.; Lukas, V.; Jaroslava, O.; Jana, H. Rapid LC-MS-based metabolomics method to study the Fusarium infection of barley. J. Sep. Sci. 2014, 37, 912–919. [Google Scholar] [CrossRef]
- Hamzehzarghani, H.; Kushalappa, A.C.; Dion, Y.; Rioux, S.; Comeau, A.; Yaylayan, V.; Marshall, W.D.; Mather, D.E. Metabolic profiling and factor analysis to discriminate quantitative resistance in wheat cultivars against fusarium head blight. Physiol. Mol. Plant Pathol. 2005, 66, 119–133. [Google Scholar] [CrossRef]
- Paranidharan, V.; Abu-Nada, Y.; Hamzehzarghani, H.; Kushalappa, A.C.; Mamer, O.; Dion, Y.; Rioux, S.; Comeau, A.; Choiniere, L. Resistance-related metabolites in wheat against Fusarium graminearum and the virulence factor deoxynivalenol (DON). Botany 2008, 86, 1168–1179. [Google Scholar] [CrossRef]
- Bollina, V.; Kumaraswamy, G.K.; Kushalappa, A.C.; Choo, T.M.; Dion, Y.; Rioux, S.; Faubert, D.; Hamzehzarghani, H. Mass spectrometry-based metabolomics application to identify quantitative resistance-related metabolites in barley against fusarium head blight. Mol. Plant Pathol. 2010, 11, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Choo, T.M. Breeding barley for resistance to fusarium head blight and mycotoxin accumulation. Plant Breed. Rev. 2010, 26, 125–169. [Google Scholar] [CrossRef]
- Kumaraswamy, K.G.; Kushalappa, A.C.; Choo, T.M.; Dion, Y.; Rioux, S. Mass spectrometry based metabolomics to identify potential biomarkers for resistance in barley against fusarium head blight (Fusarium graminearum). J. Chem. Ecol. 2011, 37, 846–856. [Google Scholar] [CrossRef]
- Talbot, N.J. On the trail of a cereal killer: Exploring the biology of Magnaporthe grisea. Annu. Rev. Microbiol. 2003, 57, 177–202. [Google Scholar] [CrossRef]
- Jones, O.A.H.; Griffin, J.L.; Jung, Y.H.; Shibato, J.; Rakwal, R.; Agrawal, G.K.; Jwa, N.S. Using metabolic profiling to assess plant-pathogen interactions: An example using rice (oryza sativa) and the blast pathogen magnaporthe grisea. Eur. J. Plant Pathol. 2011, 129, 539–554. [Google Scholar] [CrossRef]
- Manabu, N. The blast disease fungi and their metabolic products. J. Pestic. Sci. 1999, 24, 293–298. [Google Scholar]
- Jacob, S.; Grötsch, T.; Foster, A.J.; Schüffler, A.; Rieger, P.H.; Sandjo, L.P.; Liermann, J.C.; Opatz, T.; Thines, E. Unravelling the biosynthesis of pyriculol in the rice blast fungus Magnaporthe oryzae. Microbiology 2016, 163, 541–553. [Google Scholar] [CrossRef] [PubMed]
- David, P.; Manfred, B.; Hassan, Z.; Enot, D.P.; Zaira, C.R.; Overy, D.P.; Stuart, S.; Talbot, N.J.; John, D. Metabolomic analysis reveals a common pattern of metabolic re-programming during invasion of three host plant species by Magnaporthe grisea. Plant J. 2010, 59, 723–737. [Google Scholar] [CrossRef]
- Doehlemann, G.; Wahl., R.; Horst, R.J.; Voll, L.M.; Usadel, B.; Poree, F.; Stitt, M.; Pons-Kcohnemann, J.; Sonnewald, U.; Kahmann, R.; et al. Reprogramming a maize plant: Transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis. Plant J. 2010, 56, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Pan, X.; Abbas, H.M.K.; Li, F.; Dong, W. Metabolites contributing to Rhizoctonia solani AG-1-IA maturation and sclerotial differentiation revealed by UPLC-QTOF-MS metabolomics. PLoS ONE 2017, 12, e0177464. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Pan, X.; Li, F.; Dong, W. UPLC-QTOF-MS metabolomics analysis revealed the contributions of metabolites to the pathogenesis of Rhizoctonia solani strain AG-1-IA. PLoS ONE 2018, 13, e0192486. [Google Scholar] [CrossRef] [PubMed]
- Verwaaijen, B.; Wibberg, D.; Winkler, A.; Zrenner, R.; Bednarz, H.; Niehaus, K.; Grosch, R.; Pühler, A.; Schlüter, A. A comprehensive analysis of the Lactuca sativa, L. transcriptome during different stages of the compatible interaction with Rhizoctonia solani. Sci. Rep. 2019, 9, 7221. [Google Scholar] [CrossRef] [PubMed]
- Aliferis, K.A.; Faubert, D.; Jabaji, S. A metabolic profiling strategy for the dissection of plant defense against fungal pathogens. PLoS ONE 2014, 9, e111930. [Google Scholar] [CrossRef]
- Copley, T.R.; Aliferis, K.A.; Kliebenstein, D.J.; Jabaji, S.H. An integrated RNAseq-1H NMR metabolomics approach to understand soybean primary metabolism regulation in response to Rhizoctonia foliar blight disease. BMC Plant Biol. 2017, 17, 84. [Google Scholar] [CrossRef]
- Ghosh, S.; Kanwar, P.; Jha, G. Alterations in rice chloroplast integrity, photosynthesis and metabolome associated with pathogenesis of Rhizoctonia solani. Sci. Rep. 2017, 7, 41610. [Google Scholar] [CrossRef]
- Aliferis, K.A.; Jabaji, S. FT-ICR/MS and GC-EI/MS metabolomics networking unravels global potato sprout’s responses to Rhizoctonia solani infection. PLoS ONE 2012, 7, e42576. [Google Scholar] [CrossRef]
- Suharti, W.S.; Nose, A.; Zheng, S.H. Metabolomic study of two rice lines infected by Rhizoctonia solani in negative ion mode by CE/TOF-MS. J. Plant Physiol. 2016, 206, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Hayden, H.L.; Rochfort, S.J.; Ezernieks, V.; Savin, K.W.; Mele, P.M. Metabolomics approaches for the discrimination of disease suppressive soils for Rhizoctonia solani AG8 in cereal crops using 1H NMR and LC-MS. Sci. Total. Environ. 2019, 651 Pt 1, 1627–1638. [Google Scholar] [CrossRef]
- Camañes, G.; Scalschi, L.; Vicedo, B.; González-Bosch, C.; García-Agustín, P. An untargeted global metabolomic analysis reveals the biochemical changes underlying basal resistance and priming in Solanum lycopersicum, and identifies 1-methyltryptophan as a metabolite involved in plant responses to Botrytis cinerea and Pseudomonas syringae. Plant J. 2015, 84, 125–139. [Google Scholar] [CrossRef]
- Hu, Z.; Chang, X.; Dai, T.; Li, L.; Liu, P.; Wang, G.; Liu, P.; Huang, Z.; Liu, X. Metabolic profiling to identify the latent infection of strawberry by Botrytis cinerea. Evol. Bioinform. 2019, 15, 1176934319838518. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.J.; William Allwood, J.; Winder, C.L.; Dunn, W.B.; Heald, J.K.; Cristescu, S.M.; Sivakumaran, A.; Harren, F.J.; Mulema, J.; Denby, K.; et al. Metabolomic approaches reveal that cell wall modifications play a major role in ethylene-mediated resistance against Botrytis cinerea. Plant J. 2011, 67, 852–868. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Martinez, A.; Liger-Belair, G.; Jeandet, P.; Nuzillard, J.M.; Cilindre, C. Metabolomics reveals simultaneous influences of plant defence system and fungal growth in Botrytis cinerea-infected Vitis vinifera cv. Chardonnay berries. J. Exp. Bot. 2012, 63, 5773–5785. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Lovato, A.; Boscaini, F.; Salvetti, E.; Torriani, S.; Commisso, M.; Danzi, R.; Ugliano, M.; Polverari, A.; Tornielli, G.B.; et al. The induction of noble rot (Botrytis cinerea) infection during postharvest withering changes the metabolome of grapevine berries (Vitis vinifera L., cv. Garganega). Front. Plant Sci. 2017, 8, 1002. [Google Scholar] [CrossRef] [PubMed]
- Agudelo-Romero, P.; Erban, A.; Rego, C.; Carbonell-Bejerano, P.; Nascimento, T.; Sousa, L.; Martínez-Zapater, J.M.; Kopka, J.; Fortes, A.M. Transcriptome and metabolome reprogramming in Vitis vinifera cv. Trincadeira berries upon infection with Botrytis cinerea. J. Exp. Bot. 2015, 66, 1769–1785. [Google Scholar] [CrossRef]
- Robison, F.M.; Turner, M.F.; Jahn, C.E.; Schwartz, H.F.; Prenni, J.E.; Brick, M.A.; Heuberger, A.L. Common bean varieties demonstrate differential physiological and metabolic responses to the pathogenic fungus Sclerotinia sclerotiorum. Plant Cell Environ. 2018, 41, 2141–2154. [Google Scholar] [CrossRef]
- Tugizimana, F.; Djami-Tchatchou, A.T.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. Metabolomic analysis of defense-related reprogramming in Sorghum bicolor in response to Colletotrichum sublineolum infection reveals a functional metabolic web of phenylpropanoid and flavonoid pathways. Front. Plant Sci. 2019, 9, 1840. [Google Scholar] [CrossRef]
- Tugizimana, F.; Djami-Tchatchou, A.T.; Fahrmann, J.F.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. Time-resolved decoding of metabolic signatures of in vitro growth of the hemibiotrophic pathogen Colletotrichum sublineolum. Sci. Rep. 2019, 9, 3290. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.C.; Trengove, R.D.; Maker, G.L.; Oliver, R.P.; Solomon, P.S. Metabolite profiling identifies the mycotoxin alternariol in the pathogen Stagonospora nodorum. Metabolomics 2009, 5, 330–335. [Google Scholar] [CrossRef]
- Lowe, R.G.T.; Lord, M.; Rybak, K.; Trengove, R.D.; Oliver, R.P.; Solomon, P.S. Trehalose biosynthesis is involved in sporulation of Stagonospora nodorum. Fungal Genet. Biol. 2009, 46, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.; Zhang, L.; Panigrahi, P.; Dholakia, B.B.; Dewangan, V.; Chavan, S.G.; Kunjir, S.M.; Wu, X.; Li, N.; Rajmohanan, P.R.; et al. Fusarium oxysporum mediates systems metabolic reprogramming of chickpea roots as revealed by a combination of proteomics and metabolomics. Plant Biotechnol. J. 2016, 14, 1589–1603. [Google Scholar] [CrossRef]
- Kumar, Y.; Dholakia, B.B.; Panigrahi, P.; Kadoo, N.Y.; Giri, A.P.; Gupta, V.S. Metabolic profiling of chickpea-Fusarium interaction identifies differential modulation of disease resistance pathways. Phytochemistry 2015, 116, 120–129. [Google Scholar] [CrossRef]
- Buhtz, A.; Witzel, K.; Strehmel, N.; Ziegler, J.; Abel, S.; Grosch, R. Perturbations in the primary metabolism of tomato and Arabidopsis thaliana plants infected with the soil-borne fungus Verticillium dahliae. PLoS ONE 2015, 10, e0138242. [Google Scholar] [CrossRef]
- Su, X.; Lu, G.; Guo, H.; Zhang, K.; Li, X.; Cheng, H. The dynamic transcriptome and metabolomics profiling in Verticillium dahliae inoculated Arabidopsis thaliana. Sci. Rep. 2018, 8, 15404. [Google Scholar] [CrossRef]
- Sarkate, A.; Saini, S.S.; Teotia, D.; Gaid, M.; Mir, J.I.; Roy, P.; Agrawal, P.K.; Sircar, D. Comparative metabolomics of scab-resistant and susceptible apple cell cultures in response to scab fungus elicitor treatment. Sci. Rep. 2018, 8, 17844. [Google Scholar] [CrossRef] [Green Version]
- König, S.; Feussner, K.; Kaever, A.; Landesfeind, M.; Thurow, C.; Karlovsky, P.; Gatz, C.; Polle, A.; Feussner, I. Soluble phenylpropanoids are involved in the defense response of Arabidopsis against Verticillium longisporum. New Phytol. 2014, 202, 823–837. [Google Scholar] [CrossRef]
- Shinde, B.A.; Dholakia, B.B.; Hussain, K.; Panda, S.; Meir, S.; Rogachev, I.; Aharoni, A.; Giri, A.P.; Kamble, A.C. Dynamic metabolic reprogramming of steroidal glycol-alkaloid and phenylpropanoid biosynthesis may impart early blight resistance in wild tomato (Solanum arcanum Peralta). Plant Mol. Biol. 2017, 95, 411–423. [Google Scholar] [CrossRef]
- Lee, D.K.; Ahn, S.; Cho, H.Y.; Yun, H.Y.; Park, J.H.; Lim, J.; Lee, J.; Kwon, S.W. Metabolic response induced by parasitic plant-fungus interactions hinder amino sugar and nucleotide sugar metabolism in the host. Sci. Rep. 2016, 6, 37434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arens, N.; Backhaus, A.; Döll, S.; Fischer, S.; Seiffert, U.; Mock, H.P. Non-invasive presymptomatic detection of Cercospora beticola infection and identification of early metabolic responses in sugar beet. Front. Plant Sci. 2016, 7, 1377. [Google Scholar] [CrossRef] [PubMed]
- Pétriacq, P.; Stassen, J.H.; Ton, J. Spore Density determines infection strategy by the plant pathogenic fungus Plectosphaerella cucumerina. Plant Physiol. 2016, 170, 2325–2339. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Seo, M.H.; Oh, D.K.; Lee, C.H. Targeted metabolomics for Aspergillus oryzae-mediated biotransformation of soybean isoflavones, showing variations in primary metabolites. Biosci. Biotechnol. Biochem. 2014, 78, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Tao, N.; Chen, Y.; Wu, Y.; Wang, X.; Li, L.; Zhu, A. The terpene limonene induced the green mold of citrus fruit through regulation of reactive oxygen species (ROS) homeostasis in Penicillium digitatum spores. Food Chem. 2019, 277, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.J.; Kanyuka, K.; Hassani-Pak, K.; Derbyshire, M.; Andongabo, A.; Devonshire, J.; Lysenko, A.; Saqi, M.; Desai, N.M.; Powers, S.J.; et al. Transcriptome and metabolite profiling of the infection cycle of Zymoseptoria tritici on wheat reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions and a variation on the hemibiotrophic lifestyle definition. Plant Physiol. 2015, 167, 1158–1185. [Google Scholar] [CrossRef]
- Yuan, S.; Yan, J.; Wang, M.; Ding, X.; Zhang, Y.; Li, W.; Cao, J.; Jiang, W. Transcriptomic and metabolic profiling reveals ‘Green Ring’ and ‘Red Ring’ on jujube fruit upon postharvest Alternaria alternata infection. Plant Cell Physiol. 2019, 60, 844–861. [Google Scholar] [CrossRef]
- Dhokane, D.; Karre, S.; Kushalappa, A.C.; McCartney, C. Integrated metabolo-transcriptomics reveals Fusarium Head Blight candidate resistance genes in wheat QTL-Fhb2. PLoS ONE 2016, 11, e0155851. [Google Scholar] [CrossRef]
- Nussbaumer, T.; Warth, B.; Sharma, S.; Ametz, C.; Bueschl, C.; Parich, A.; Pfeifer, M.; Siegwart, G.; Steiner, B.; Lemmens, M.; et al. Joint Transcriptomic and metabolomic analyses Reveal Changes in the Primary Metabolism and Imbalances in the Subgenome Orchestration in the Bread wheat molecular response to Fusarium graminearum. Genes Genomes Genet. 2015, 5, 2579–2592. [Google Scholar] [CrossRef]
- Gunnaiah, R.; Kushalappa, A.C.; Duggavathi, R.; Fox, S.; Somers, D.J. Integrated metabolo-proteomic approach to decipher the mechanisms by which wheat QTL (Fhb1) contributes to resistance against Fusarium graminearum. PLoS ONE 2012, 7, e40695. [Google Scholar] [CrossRef]
- Ghosh, S.; Narula, K.; Sinha, A.; Ghosh, R.; Jawa, P.; Chakraborty, N.; Chakraborty, S. Proteometabolomic analysis of transgenic tomato overexpressing oxalate decarboxylase uncovers novel proteins potentially involved in defense mechanism against Sclerotinia. J. Proteomics 2016, 143, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Singh, M.; Pandey, D.; Kumar, A. Integrated proteomics, genomics, metabolomics approaches reveal oxalic acid as pathogenicity factor in Tilletia indica inciting Karnal bunt disease of wheat. Sci. Rep. 2018, 8, 7826. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.R.; Wang, Q.; Li, B.; Friedt, W.; Römpp, A.; Spengler, B.; Gottwald, S. Histology-guided high-resolution AP-SMALDI mass spectrometry imaging of wheat-Fusarium graminearum interaction at the root-shoot junction. Plant Methods 2018, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Gunnaiah, R.; Kushalappa, A.C. Metabolomics deciphers the host resistance mechanisms in wheat cultivar Sumai-3, against trichothecene producing and non-producing isolates of Fusarium graminearum. Plant Physiol. Biochem. 2014, 83, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Kumaraswamy, G.K.; Kushalappa, A.C.; Choo, T.M.; Dion, Y.; Rioux, S. Differential metabolic response of barley genotypes, varying in resistance, to trichothecene-producing and -nonproducing (tri5−) isolates of Fusarium graminearum. Plant Pathol. 2011, 61, 509–521. [Google Scholar] [CrossRef]
- Scandiani, M.M.; Luque, A.G.; Razori, M.V.; Ciancio Casalini, L.; Aoki, T.; O’Donnell, K.; Cervigni, G.D.; Spampinato, C.P. Metabolic profiles of soybean roots during early stages of Fusarium tucumaniae infection. J. Exp. Bot. 2015, 66, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Westrick, N.M.; Jain, S.; Piotrowski, J.S.; Ranjan, M.; Kessens, R.; Stiegman, L.; Grau, C.R.; Conley, S.P.; Smith, D.L.; et al. Resistance against Sclerotinia sclerotiorum in soybean involves a reprogramming of the phenylpropanoid pathway and up-regulation of antifungal activity targeting ergosterol biosynthesis. Plant Biotechnol. J. 2019, 17, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Wojakowska, A.; Muth, D.; Narożna, D.; Mądrzak, C.; Stobiecki, M.; Kachlicki, P. Changes of phenolic secondary metabolite profiles in the reaction of narrow leaf lupin (Lupinus angustifolius) plants to infections with Colletotrichum lupini fungus or treatment with its toxin. Metabolomics 2013, 9, 575–589. [Google Scholar] [CrossRef]
- Botanga, C.J.; Bethke, G.; Chen, Z.; Gallie, D.R.; Fiehn, O.; Glazebrook, J. Metabolite profiling of Arabidopsis inoculated with Alternaria brassicicola reveals that ascorbate reduces disease severity. Mol. Plant Microbe Interact. 2012, 25, 1628–1638. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| NO | Name | Website Address |
|---|---|---|
| 1 | ECMDB: The Escherichia coli Metabolome Database | http://www.ecmdb.ca/ |
| 2 | YMDB: The Yeast Metabolome Database | http://www.ymdb.ca/ |
| 3 | HMP: The Human Microbiome Project | http://www.hmpdacc.org/ |
| 4 | EcoCyc: Encyclopedia of Escherichia coli K-12 Genes and Metabolism | http://www.ecocyc.org/ |
| 5 | NMD: National Microbiological Database | http://www.foodsafety.govt.nz/industry/general/nmd/ |
| 6 | MNPD: Microbial Natural Products Database | http://naturalprod.ucsd.edu/ |
| 7 | UMBBD: University of Minnesota Biocatalysis/Biodegradation Database | http://umbbd.ethz.ch/ |
| 8 | BioCyc Pathway | http://biocyc.org/ |
| 9 | HMDB: Human Metabolome Database | http://www.hmdb.ca/ |
| 10 | KEGG: Kyoto Encyclopedia of Genes and Genomes | http://www.genome.jp/kegg/ |
| 11 | HumanCyc | http://bicyc.org |
| 12 | ARM | http://www.metabolome.jp |
| 13 | Lipidomics: Lipid Maps | http://www.lipidmaps.org/data/index.html |
| 14 | Lipidomics: SphinGOMAP | http://sphingomap.org/ |
| 15 | Lipidomics: Lipid Bank | http://lipidbank.jp/ |
| 16 | New drug and its metabolite database | http://www.ualberta.ca/_gjones/mslib.htm |
| 17 | ChemSpider Beta | http://www.chemspider.com |
| 18 | METLIN | http://metlin.scripps.edu/ |
| 19 | MetaCyc Encyclopedia of Metabolic Pathways | http://metacyc.org/ |
| 20 | PubChem Compound | http://www.pubmed.gov |
| 21 | SYSTOMONAS genome Database | http://systomonas.tu-bs.de/ |
| 22 | PathDB: Pathogen Database | http://www.ncgr.org/pathdb/ |
| 23 | NIST: National Institute of Standards and Technology | http://www.NIST.gov/srd/ |
| Fungal Pathogen | Plant Host | Platform | Year [Ref] |
|---|---|---|---|
| Fusarium graminearum | wheat | AP-SMALDI-MS | 2018 [103] |
| wheat | LC-ESI-LTQ-Orbitrap | 2014 [104] | |
| barley | UHPLC-MS/MS | 2014 [52]; 2011 [12] | |
| Arabidopsis | 1H NMR | 2018 [44] | |
| barley | LC-ESI-LTQ-Orbitrap | 2012 [105]; 2010 [55] | |
| Fusarium oxysporum | chickpea | 1H NMR | 2016 [84] |
| chickpea | UHPLC-ESI-MS/MS | 2015 [85] | |
| Fusarium tucumaniae | soybean | GC-MS | 2015 [106] |
| Magnaporthe oryzae | barley and rice | GC-MS | 2009 [62] |
| rice | 1H NMR, LC-MS and GC-MS | 2011 [58] | |
| rice | LC-MS and 1H NMR | 2016 [61] | |
| Ustilago maydis | maize | LC-MS | 2008 [63] |
| Rhizoctonia solani | rice | UPLC-QTOF-MS | 2017 [64]; 2018 [65] |
| wheat and barley | 1H NMR and LC-MS | 2019 [72] | |
| rice | GC-MS and CE/TOF-MS | 2017 [69]; 2016 [71] | |
| soybean | GC-MS | 2014 [67] | |
| soybean | 1H NMR | 2017 [68] | |
| lettuce | GC-MS | 2019 [66] | |
| potato | FT-ICR/MS and GC-EI/MS | 2012 [70] | |
| Botrytis cinerea | tomato | LC-MS and GC-MS | 2015 [73] |
| strawberry | GC-MS | 2019 [74] | |
| Arabidopsis | DI-MS | 2011 [75] | |
| grape | GC-MS | 2017 [77]; 2015 [78] | |
| grape | 1H NMR | 2012 [76]; | |
| Sclerotinia sclerotiorum | common bean | UPLC-MS and GC-MS | 2018 [79] |
| tomato | UPLC-QTOF-MS/MS | 2016 [101] | |
| soybean | GC-MS | 2019 [107] | |
| Colletotrichum lupini | lupin | LC-MS and GC-MS | 2013 [108] |
| Colletotrichum sublineolum | sorghum | LC-ESI-QTOF-MS | 2019 [80] |
| sorghum | UHPLC-QTOF-MS | 2019 [81] | |
| Verticillium dahliae | Arabidopsis | GC-MS and LC-ESI-MS/MS | 2015 [86] |
| Arabidopsis | 1H NMR | 2018 [87] | |
| Verticillium longisporum | Arabidopsis | UHPLC-QTOF-MS | 2014 [89] |
| Venturia inaequalis | apple | GC-MS | 2018 [88] |
| Alternaria solani | wild tomato | UPLC-QTOF-MS/LC-MS | 2017 [90] |
| Alternaria brassicicola | Arabidopsis | GC-MS | 2012 [109] |
| Gymnosporangium asiaticum | Rosaceae plants | GC-MS | 2016 [91] |
| Cercospora beticola | sugar beet | (U)HPLC-UV-ESI-MS | 2016 [92] |
| Plectosphaerella cucumerina | Arabidopsis | UPLC-QTOF-MS/MS | 2016 [93] |
| Aspergillus oryzae | soybean | LC-ESI-MS and GC-TOF-MS | 2014 [94] |
| Penicillium digitatum | citrus | GC–MS | 2018 [95] |
| Zymoseptoria tritici | wheat | UHLC-MS/MS and GC-MS | 2015 [96] |
| Stagonospora nodorum | wheat | GC-MS and ESI-MS/MS | 2009 [82] |
| Alternaria alternata | jujube fruit | UPLC-QTOF-MS/MS | 2019 [97] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Ma, R.; Chen, X.-L. Advances of Metabolomics in Fungal Pathogen–Plant Interactions. Metabolites 2019, 9, 169. https://doi.org/10.3390/metabo9080169
Chen F, Ma R, Chen X-L. Advances of Metabolomics in Fungal Pathogen–Plant Interactions. Metabolites. 2019; 9(8):169. https://doi.org/10.3390/metabo9080169
Chicago/Turabian StyleChen, Fangfang, Ruijing Ma, and Xiao-Lin Chen. 2019. "Advances of Metabolomics in Fungal Pathogen–Plant Interactions" Metabolites 9, no. 8: 169. https://doi.org/10.3390/metabo9080169
APA StyleChen, F., Ma, R., & Chen, X. -L. (2019). Advances of Metabolomics in Fungal Pathogen–Plant Interactions. Metabolites, 9(8), 169. https://doi.org/10.3390/metabo9080169