
Constraints on the Production of Phosphine by Venusian Volcanoes

 , ,
, ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phosphorus Partition between Iron and Silicate
2.2. Phosphorus Thermodynamics in Silicate Melts
2.3. Phosphorus and Oxygen Diffusion through Solids
3. Results
3.1. Eruptive Melt Minerology Is Determined in the Upper Mantle and Lithosphere
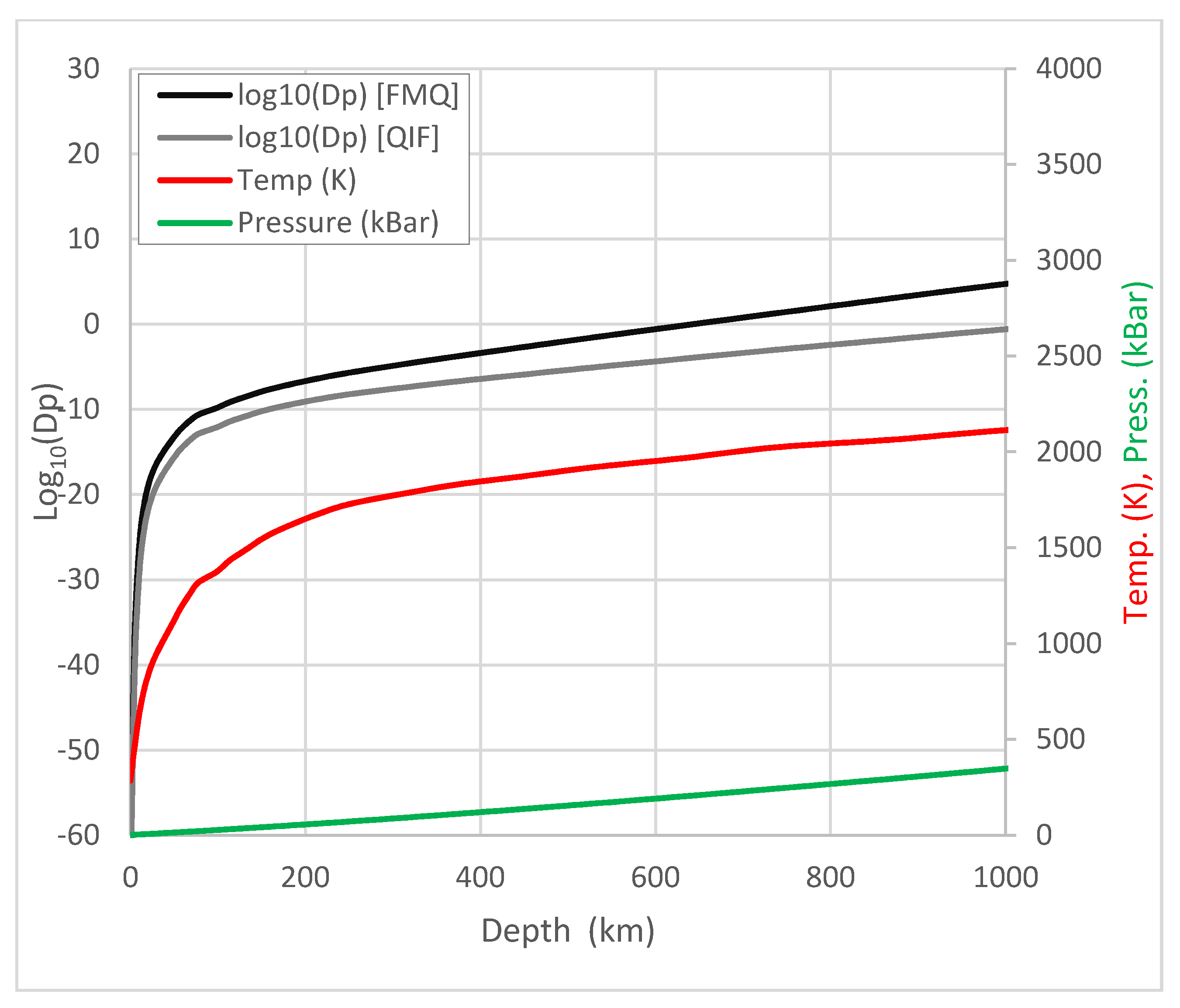
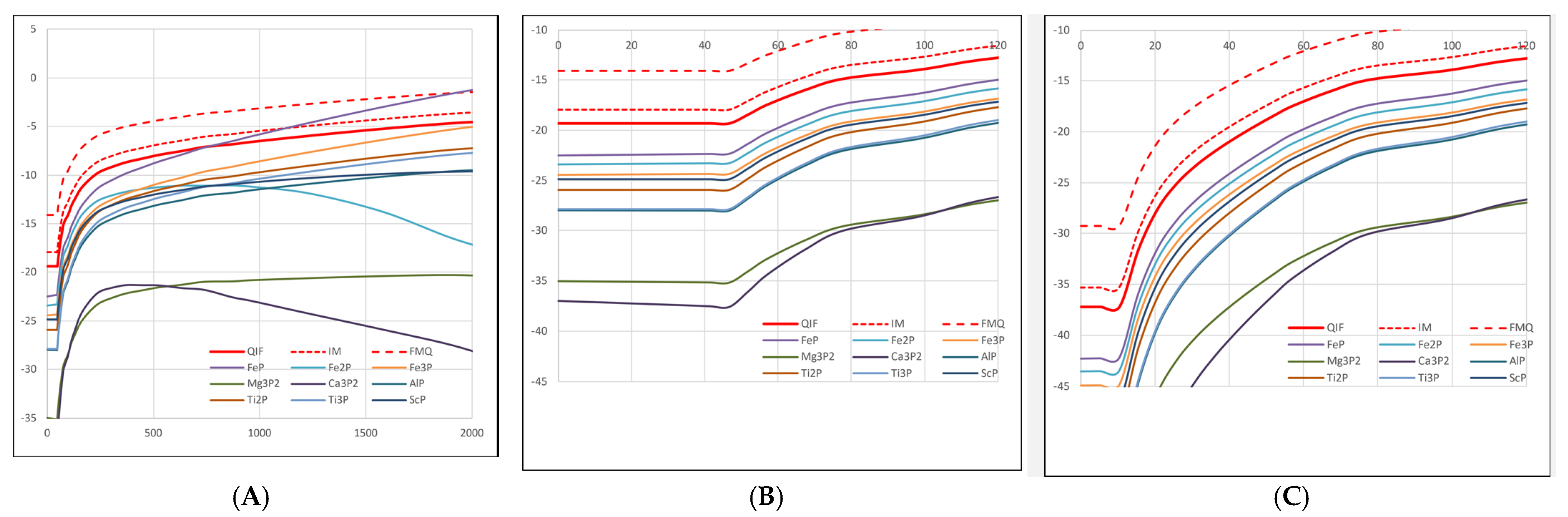
3.2. Phosphorus Redox State in the Lithosphere and Upper Mantle
3.3. Mechanisms of Explosive Volcanism on Venus
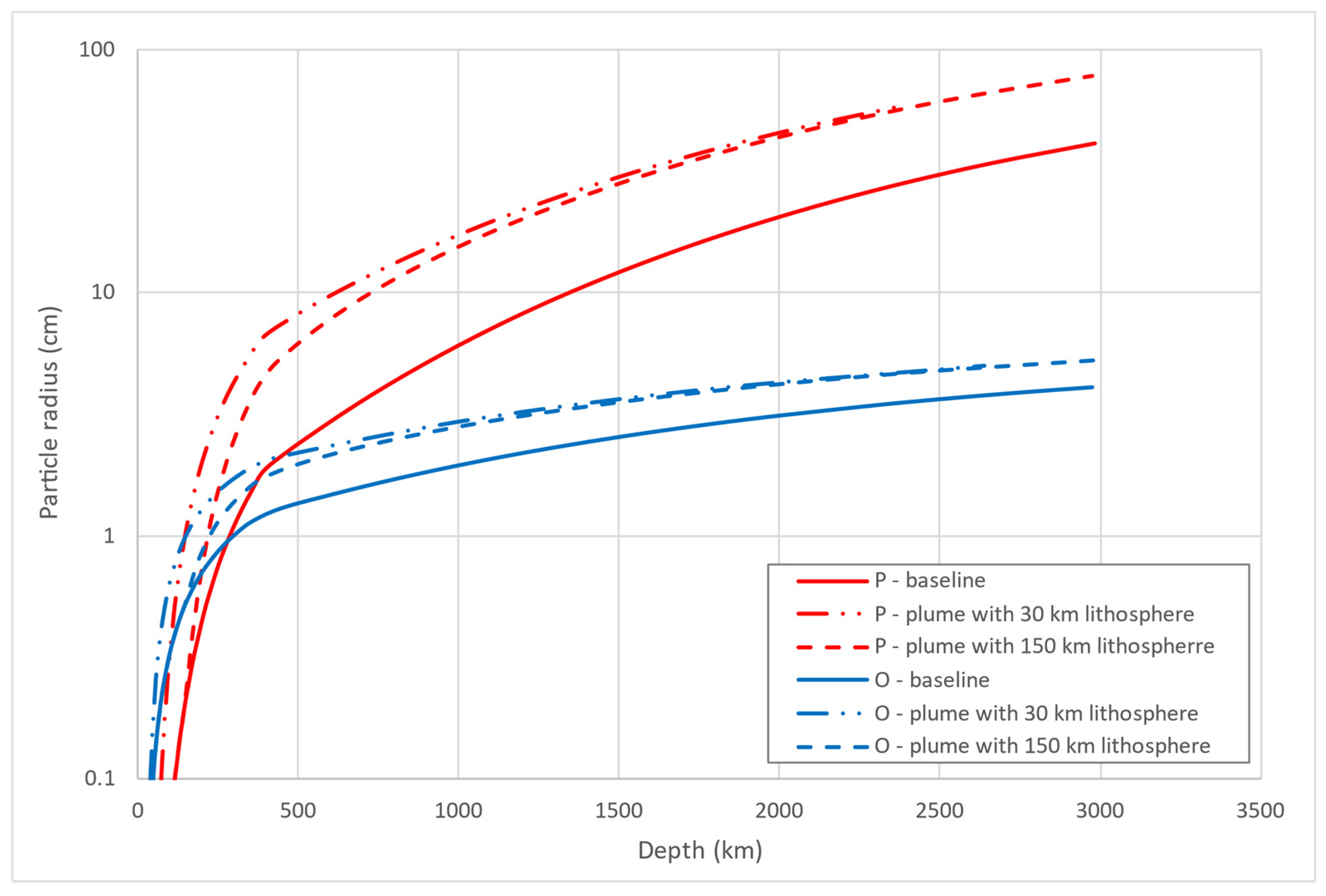
3.4. Ash Plume Generation on Venus
3.5. Other Criteria for a Volcanic Source
3.6. Required Rate of Phosphide Volcanic Eruption
- It assumes that phosphine is only present from 53–61 km. However, the very same diffusive processes invoked to transport phosphine to the upper atmospheres mean that phosphine must be present at altitudes beyond 53–61 km as well. In other words, this approach underestimates .
- It overestimates the lifetime of high-altitude phosphine. The eddy diffusion coefficient increases with altitude in the Venusian atmosphere, meaning that the use of 103 cm2 s−1 underestimates the phosphine destruction rate. Therefore, it overestimates for PH3 at z > 63 km, which must exist due to the diffusive transport.
- It assumes that phosphine is only destroyed after transport. In fact, phosphine is continually destroyed throughout the Venusian atmosphere due to radical sinks [1].
4. Discussion
- (a)
- The atmospheric structure and gas transport on Venus are such that PH3 (and only PH3) is retained solely in the lower cloud layer, and does not diffuse upwards or downwards, and
- (b)
- The Venusian lithosphere is substantially more reduced than is expected from lander data and atmospheric chemistry, and
- (c)
- Volcanism is currently erupting tens of thousands of cubic kilometers of magma onto the surface per year, and
- (d)
- Venusian volcanism is unexpectedly efficient at generating high altitude clouds of fine magma ash.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greaves, J.S.; Richards, A.M.; Bains, W.; Rimmer, P.B.; Sagawa, H.; Clements, D.L.; Seager, S.; Petkowski, J.J.; Sousa-Silva, C.; Ranjan, S. Phosphine gas in the cloud decks of Venus. Nat. Astron. 2020, 5, 655–664. [Google Scholar] [CrossRef]
- Greaves, J.S.; Richards, A.M.S.; Bains, W.; Rimmer, P.B.; Sagawa, H.; Clements, D.L.; Seager, S.; Petkowski, J.J.; Sousa-Silva, C.; Ranjan, S.; et al. Addendum: Phosphine gas in the cloud deck of Venus. Nat. Astron. 2021, 5, 726–728. [Google Scholar] [CrossRef]
- Trompet, L.; Robert, S.; Mahieux, A.; Schmidt, F.; Erwin, J.; Vandaele, A. Phosphine in Venus’ atmosphere: Detection attempts and upper limits above the cloud top assessed from the SOIR/VEx spectra. Astron. Astrophys. 2021, 645, L4. [Google Scholar] [CrossRef]
- Villanueva, G.L.; Cordiner, M.; Irwin, P.G.J.; de Pater, I.; Butler, B.; Gurwell, M.; Milam, S.N.; Nixon, C.A.; Luszcz-Cook, S.H.; Wilson, C.F.; et al. No evidence of phosphine in the atmosphere of Venus from independent analyses. Nat. Astron. 2021, 5, 631–635. [Google Scholar] [CrossRef]
- Akins, A.B.; Lincowski, A.P.; Meadows, V.S.; Steffes, P.G. Complications in the ALMA detection of phosphine at Venus. Astrophys. J. 2021, 907, L27. [Google Scholar] [CrossRef]
- Lincowski, A.P.; Meadows, V.S.; Crisp, D.; Akins, A.B.; Schwieterman, E.W.; Arney, G.N.; Wong, M.L.; Steffes, P.G.; Parenteau, M.N.; Domagal-Goldman, S. Claimed detection of PH3 in the clouds of Venus is consistent with mesospheric SO2. Astrophys. J. Lett. 2021, 908, L44. [Google Scholar] [CrossRef]
- Greaves, J.S.; Rimmer, P.B.; Richards, A.; Petkowski, J.J.; Bains, W.; Ranjan, S.; Seager, S.; Clements, D.L.; Silva, C.S.; Fraser, H.J. Low Levels of sulphur dioxide contamination of phosphine spectra from Venus’ atmosphere. arXiv 2021, arXiv:2108.08393. [Google Scholar]
- Greaves, J.S.; Richards, A.M.; Bains, W.; Rimmer, P.B.; Clements, D.L.; Seager, S.; Petkowski, J.J.; Sousa-Silva, C.; Ranjan, S.; Fraser, H.J. Reply to: No evidence of phosphine in the atmosphere of Venus from independent analyses. Nat. Astron. 2021, 5, 636–639. [Google Scholar] [CrossRef]
- Mogul, R.; Limaye, S.S.; Way, M.J.; Cordova, J.A. Venus’ mass spectra show signs of disequilibria in the middle clouds. Geophys. Res. Lett. 2021, 48, e2020GL091327. [Google Scholar] [CrossRef]
- Omran, A.; Oze, C.; Jackson, B.; Mehta, C.; Barge, L.M.; Bada, J.; Pasek, M.A. Phosphine generation pathways on rocky planets. Astrobiology 2021, 21, 1264–1276. [Google Scholar] [CrossRef]
- Bains, W.; Petkowski, J.J.; Seager, S.; Ranjan, S.; Sousa-Silva, C.; Rimmer, P.B.; Zhan, Z.; Greaves, J.S.; Richards, A.M. Phosphine on Venus cannot be explained by conventional processes. Astrobiology 2021, 21, 1277–1304. [Google Scholar] [CrossRef] [PubMed]
- Bains, W.; Petkowski, J.J.; Seager, S.; Ranjan, S.; Sousa-Silva, C.; Rimmer, P.B.; Zhan, Z.; Greaves, J.S.; Richards, A.M. Venusian phosphine: A ‘wow!’signal in chemistry? In Phosphorus, Sulfur, and Silicon and the Related Elements; Taylor & Francis: London, UK, 2021; pp. 1–6. [Google Scholar]
- Bains, W.; Petkowski, J.J.; Zhan, Z.; Seager, S. Evaluating alternatives to water as solvents for life: The example of sulfuric acid. Life 2021, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, P.B.; Jordan, S.; Constantinou, T.; Woitke, P.; Shorttle, O.; Hobbs, R.; Paschodimas, A. Hydroxide salts in the clouds of Venus: Their effect on the sulfur cycle and cloud droplet pH. Planet. Sci. J. 2021, 2, 133. [Google Scholar] [CrossRef]
- Bains, W.; Petkowski, J.J.; Rimmer, P.B.; Seager, S. Production of ammonia makes venusian clouds habitable and explains observed cloud-level chemical anomalies. Proc. Natl. Acad. Sci. USA 2021, 118, e2110889118. [Google Scholar] [CrossRef]
- Truong, N.; Lunine, J. Volcanically extruded phosphides as an abiotic source of Venusian phosphine. Proc. Natl. Acad. Sci. USA 2021, 118, e2021689118. [Google Scholar] [CrossRef]
- Strom, R.G.; Schaber, G.G.; Dawson, D.D. The global resurfacing of Venus. J. Geophys. Res. Planets 1994, 99, 10899–10926. [Google Scholar] [CrossRef]
- Gu, T.; Stagno, V.; Fei, Y. Partition coefficient of phosphorus between liquid metal and silicate melt with implications for the Martian magma ocean. Phys. Earth Planet. Inter. 2019, 295, 106298. [Google Scholar] [CrossRef]
- Dziewonski, A.M.; Anderson, D.L. Preliminary reference Earth model. Phys. Earth Planet. Inter. 1981, 25, 297–356. [Google Scholar] [CrossRef]
- Spiliopoulos, S.; Stacey, F.D. The earth’s thermal profile: Is there a mid-mantle thermal boundary layer? J. Geodyn. 1984, 1, 61–77. [Google Scholar] [CrossRef]
- Dannberg, J.; Sobolev, S.V. Low-buoyancy thermochemical plumes resolve controversy of classical mantle plume concept. Nat. Commun. 2015, 6, 6960. [Google Scholar] [CrossRef] [Green Version]
- Chase, M.W.J. NIST-JANAF thermochemical tables. J. Phys. Chem. Ref. Data 1998, 9. [Google Scholar] [CrossRef]
- Robie, R.A.; Hemingway, B.S. Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 bar (105 Pascals) Pressure and at Higher Temperatures; US Government Printing Office: Washington, DC, USA, 1995.
- Zaitsev, A.; Dobrokhotova, Z.V.; Litvina, A.; Mogutnov, B. Thermodynamic properties and phase equilibria in the Fe–P system. J. Chem. Soc. Faraday Trans. 1995, 91, 703–712. [Google Scholar] [CrossRef]
- Barin, I. (Ed.) Thermochemical Data of Pure Substances, 3rd ed.; VCH Verlagsgesellschaft mbH: Berlin, Germany, 1995. [Google Scholar]
- Schlesinger, M.E. The Thermodynamic Properties of Phosphorus and Solid Binary Phosphides. Chem. Rev. 2002, 102, 4267–4302. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Frost, B.R. Introduction to oxygen fugacity and its petrologic significance. In Reviews in Minerology Volume 25. Oxide Minerals: Petrologic and Magnetic Significance; Lindsleys, D.H., Banerjee, S.K., Eds.; Minerological Society of America: Washington, DC, USA, 1991; pp. 1–10. [Google Scholar]
- Uemura, K. Diffusion of elements in solid iron and steel. Tetsu-to-Hagane 1943, 29, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Posselt, M.; Faßbender, J. Influence of substitutional atoms on the diffusion of oxygen in dilute iron alloys. Phys. Rev. B 2018, 98, 064103. [Google Scholar] [CrossRef]
- Stolper, E.M.; Shorttle, O.; Antoshechkina, P.M.; Asimow, P.D. The effects of solid-solid phase equilibria on the oxygen fugacity of the upper mantle. Am. Mineral. J. Earth Planet. Mater. 2020, 105, 1445–1471. [Google Scholar] [CrossRef]
- Griffin, W.; Gain, S.; Cámara, F.; Bindi, L.; Shaw, J.; Alard, O.; Saunders, M.; Huang, J.-X.; Toledo, V.; O’Reilly, S. Extreme reduction: Mantle-derived oxide xenoliths from a hydrogen-rich environment. Lithos 2020, 358, 105404. [Google Scholar] [CrossRef]
- Choudhuri, M.; Nemčok, M. Plumes and Hotspots. In Mantle Plumes and Their Effects; Choudhuri, M., Nemčok, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 19–42. [Google Scholar]
- Sleep, N.H. Hotspot volcanism and mantle plumes. Ann. Rev. Earth Planet. Sci. 1992, 20, 19–43. [Google Scholar] [CrossRef]
- Loper, D.E. Mantle plumes and their effect on the Earth’s surface: A review and synthesis. Dyn. Atmos. Ocean. 1998, 27, 35–54. [Google Scholar] [CrossRef]
- Sleep, N.H.; Ebinger, C.J.; Kendall, J.-M. Deflection of mantle plume material by cratonic keels. Geol. Soc. Lond. Spec. Publ. 2002, 199, 135–150. [Google Scholar] [CrossRef]
- Williams, Q.; Jeanloz, R.; Bass, J.; Svendsen, B.; Ahrens, T.J. The melting curve of iron to 250 gigapascals: A constraint on the temperature at earth’s center. Science 1987, 236, 181–182. [Google Scholar] [CrossRef] [PubMed]
- d’Acremont, E.; Leroy, S.; Burov, E.B. Numerical modelling of a mantle plume: The plume head–lithosphere interaction in the formation of an oceanic large igneous province. Earth Planet. Sci. Lett. 2003, 206, 379–396. [Google Scholar] [CrossRef]
- Smrekar, S.; Auerbach, V.; Ostberg, C.; O’Rourke, J. How thick is the lithosphere of Venus? LPI Contrib. 2019, 2193, 8035. [Google Scholar]
- Uppalapati, S.; Rolf, T.; Crameri, F.; Werner, S.C. Dynamics of lithospheric overturns and implications for Venus’s surface. J. Geophys. Res. Planets 2020, 125, e2019JE006258. [Google Scholar] [CrossRef]
- Behn, M.D.; Hirth, G.; Elsenbeck, J.R. Implications of grain size evolution on the seismic structure of the oceanic upper mantle. Earth Planet. Sci. Lett. 2009, 282, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Dannberg, J.; Eilon, Z.; Faul, U.; Gassmöller, R.; Moulik, P.; Myhill, R. The importance of grain size to mantle dynamics and seismological observations. Geochem. Geophys. Geosyst. 2017, 18, 3034–3061. [Google Scholar] [CrossRef]
- Mazza, S.E.; Gazel, E.; Bizimis, M.; Moucha, R.; Béguelin, P.; Johnson, E.A.; McAleer, R.J.; Sobolev, A.V. Sampling the volatile-rich transition zone beneath Bermuda. Nature 2019, 569, 398–403. [Google Scholar] [CrossRef]
- Beyer, C.; Frost, D.J. The depth of sub-lithospheric diamond formation and the redistribution of carbon in the deep mantle. Earth Planet. Sci. Lett. 2017, 461, 30–39. [Google Scholar] [CrossRef]
- Kaminsky, F.V.; Wirth, R.; Schreiber, A. Carbonatitic inclusions in deep mantle diamond from Juina, Brazil: New minerals in the carbonate-halide association. Can. Mineral. 2013, 51, 669–688. [Google Scholar] [CrossRef]
- Palot, M.; Pearson, D.G.; Stern, R.A.; Stachel, T.; Harris, J.W. Isotopic constraints on the nature and circulation of deep mantle C–H–O–N fluids: Carbon and nitrogen systematics within ultra-deep diamonds from Kankan (Guinea). Geochim. Cosmochim. Acta 2014, 139, 26–46. [Google Scholar] [CrossRef]
- Pearson, D.; Brenker, F.; Nestola, F.; McNeill, J.; Nasdala, L.; Hutchison, M.; Matveev, S.; Mather, K.; Silversmit, G.; Schmitz, S. Hydrous mantle transition zone indicated by ringwoodite included within diamond. Nature 2014, 507, 221–224. [Google Scholar] [CrossRef]
- Smith, E.M.; Shirey, S.B.; Nestola, F.; Bullock, E.S.; Wang, J.; Richardson, S.H.; Wang, W. Large gem diamonds from metallic liquid in Earth’s deep mantle. Science 2016, 354, 1403–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschauner, O.; Huang, S.; Greenberg, E.; Prakapenka, V.; Ma, C.; Rossman, G.; Shen, A.; Zhang, D.; Newville, M.; Lanzirotti, A. Ice-VII inclusions in diamonds: Evidence for aqueous fluid in Earth’s deep mantle. Science 2018, 359, 1136–1139. [Google Scholar] [CrossRef] [Green Version]
- Scott, E.R. Iron meteorites: Composition, age, and origin. In Oxford Research Encyclopedia of Planetary Science; Oxford University Press: Oxford, UK, 2020. [Google Scholar]
- Ulff-Moller, F. Solidification history of the Kitdlit Lens: Immiscible metal and sulphide liquids from a basaltic dyke on Disko, central West Greenland. J. Petrol. 1985, 26, 64–91. [Google Scholar] [CrossRef]
- Xiong, Q.; Griffin, W.L.; Huang, J.-X.; Gain, S.E.; Toledo, V.; Pearson, N.J.; O’Reilly, S.Y. Super-reduced mineral assemblages in “ophiolitic” chromitites and peridotites: The view from Mount Carmel. Eur. J. Mineral. 2017, 29, 557–570. [Google Scholar] [CrossRef]
- Surkov, Y.A.; Barsukov, V.L.; Moskalyeva, L.P.; Kharyukova, V.P.; Kemurdzhian, A.L. New data on the composition, structure, and properties of Venus rock obtained by Venera 13 and Venera 14. J. Geophys. Res. Solid Earth 1984, 89, B393–B402. [Google Scholar] [CrossRef]
- Surkov, Y.A.; Moskalyova, L.P.; Kharyukova, V.P.; Dudin, A.D.; Smirnov, G.G.; Zaitseva, S.Y. Venus rock composition at the Vega 2 Landing Site. J. Geophys. Res. Solid Earth 1986, 91, E215–E218. [Google Scholar] [CrossRef]
- Gilmore, M.S.; Mueller, N.; Helbert, J. VIRTIS emissivity of Alpha Regio, Venus, with implications for tessera composition. Icarus 2015, 254, 350–361. [Google Scholar] [CrossRef]
- Fegley, B. 2.7–Venus. In Treatise on Geochemistry, 2nd ed.; Holland, H.D., Turekian, K.K., Eds.; Elsevier: Oxford, UK, 2014; pp. 127–148. [Google Scholar]
- Fegley, B.; Prinn, R.G. Estimation of the rate of volcanism on Venus from reaction rate measurements. Nature 1989, 337, 55–58. [Google Scholar] [CrossRef]
- Krasnopolsky, V.A. High-resolution spectroscopy of Venus: Detection of OCS, upper limit to H2S, and latitudinal variations of CO and HF in the upper cloud layer. Icarus 2008, 197, 377–385. [Google Scholar] [CrossRef]
- Yin, Y.; Zhai, K.; Zhang, B.; Zhai, S. Electrical resistivity of iron phosphides at high-pressure and high-temperature conditions with implications for lunar core’s thermal conductivity. J. Geophys. Res. Solid Earth 2019, 124, 5544–5556. [Google Scholar] [CrossRef]
- Pearson, D.G.; Scott, J.M.; Liu, J.; Schaeffer, A.; Wang, L.H.; van Hunen, J.; Szilas, K.; Chacko, T.; Kelemen, P.B. Deep continental roots and cratons. Nature 2021, 596, 199–210. [Google Scholar] [CrossRef]
- Leung, I.; Guo, W.; Friedman, I.; Gleason, J. Natural occurrence of silicon carbide in a diamondiferous kimberlite from Fuxian. Nature 1990, 346, 352–354. [Google Scholar] [CrossRef]
- Robinson, P.T.; Bai, W.-J.; Malpas, J.; Yang, J.-S.; Zhou, M.-F.; Fang, Q.-S.; Hu, X.-F.; Cameron, S.; Staudigel, H. Ultra-high pressure minerals in the Luobusa Ophiolite, Tibet, and their tectonic implications. Geol. Soc. Lond. Spec. Publ. 2004, 226, 247–271. [Google Scholar] [CrossRef]
- Pierro, S.D.; Gnos, E.; Grobéty, B.; Armbruster, T.M.; Bernasconi, S.; Ulmer, P. Rock-forming moissanite (natural ∂-silicon carbide). Am. Mineral. 2003, 88, 1817–1821. [Google Scholar] [CrossRef]
- Yang, J.; Meng, F.; Xu, X.; Robinson, P.T.; Dilek, Y.; Makeyev, A.B.; Wirth, R.; Wiedenbeck, M.; Cliff, J. Diamonds, native elements and metal alloys from chromitites of the Ray-Iz ophiolite of the Polar Urals. Gondwana Res. 2015, 27, 459–485. [Google Scholar] [CrossRef]
- Shiryaev, A.A.; Griffin, W.L.; Stoyanov, E. Moissanite (SiC) from kimberlites: Polytypes, trace elements, inclusions and speculations on origin. Lithos 2011, 122, 152–164. [Google Scholar] [CrossRef]
- Xu, S.; Wu, W.; Xiao, W.; Yang, J.; Chen, J.; Ji, S.; Yican Liu, U. Moissanite in serpentinite from the Dabie Mountains in China. Mineral. Mag. 2018, 72, 899–908. [Google Scholar] [CrossRef]
- Airey, M.W.; Mather, T.; Pyle, D.; Glaze, L.; Ghail, R.; Wilson, C. Explosive volcanic activity on Venus: The roles of volatile contribution, degassing, and external environment. Planet. Space Sci. 2015, 113, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Dera, P.; Lavina, B.; Borkowski, L.A.; Prakapenka, V.B.; Sutton, S.R.; Rivers, M.L.; Downs, R.T.; Boctor, N.Z.; Prewitt, C.T. High-pressure polymorphism of Fe2P and its implications for meteorites and Earth’s core. Geophys. Res. Lett. 2008, 35, L10301. [Google Scholar] [CrossRef] [Green Version]
- Britvin, S.; Murashko, M.; Vapnik, E.; Polekhovsky, Y.S.; Krivovichev, S. Barringerite Fe2P from pyrometamorphic rocks of the Hatrurim Formation, Israel. Geol. Ore Depos. 2017, 59, 619–625. [Google Scholar] [CrossRef]
- Krasnopolsky, V.A. Chemical kinetic model for the lower atmosphere of Venus. Icarus 2007, 191, 25–37. [Google Scholar] [CrossRef]
- Glaze, L.S. Transport of SO2 by explosive volcanism on Venus. J. Geophys. Res. Planets 1999, 104, 18899–18906. [Google Scholar] [CrossRef]
- Esposito, L.W. Sulfur dioxide: Episodic injection shows evidence for active Venus volcanism. Science 1984, 223, 1072–1074. [Google Scholar] [CrossRef]
- La Femina, P.C.; Connor, C.B.; Hill, B.E.; Strauch, W.; Saballos, J.A. Magma–tectonic interactions in Nicaragua: The 1999 seismic swarm and eruption of Cerro Negro volcano. J. Volcanol. Geotherm. Res. 2004, 137, 187–199. [Google Scholar] [CrossRef]
- Carr, M.; Feigenson, M.; Patino, L.; Walker, J. Volcanism and geochemistry in Central America: Progress and problems. Geophys. Monogr.-Am. Geophys. Union 2003, 138, 153–174. [Google Scholar]
- Cole, J.W. Structure, petrology, and genesis of Cenozoic volcanism, Taupo Volcanic Zone, New Zealand—A review. N. Zeal. J. Geol. Geophys. 1979, 22, 631–657. [Google Scholar] [CrossRef] [Green Version]
- Hogg, A.G.; Higham, T.F.; Lowe, D.J.; Palmer, J.G.; Reimer, P.J.; Newnham, R.M. A wiggle-match date for Polynesian settlement of New Zealand. Antiquity 2003, 77, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Sahetapy-Engel, S.; Self, S.; Carey, R.J.; Nairn, I.A. Deposition and generation of multiple widespread fall units from the c. AD 1314 Kaharoa rhyolitic eruption, Tarawera, New Zealand. Bull. Volcanol. 2014, 76, 836. [Google Scholar] [CrossRef]
- Colgate, S.A.; Sigurgeisson, T. Dynamic mixing of water and lava. Nature 1973, 244, 552–555. [Google Scholar] [CrossRef]
- Ort, M.H.; Di Muro, A.; Michon, L.; Bachèlery, P. Explosive eruptions from the interaction of magmatic and hydrothermal systems during flank extension: The Bellecombe Tephra of Piton de La Fournaise (La Réunion Island). Bull. Volcanol. 2016, 78, 5. [Google Scholar] [CrossRef] [Green Version]
- Antonopoulos, J. The great Minoan eruption of Thera volcano and the ensuing tsunami in the Greek Archipelago. Nat. Hazards 1992, 5, 153–168. [Google Scholar] [CrossRef]
- Bachmann, O.; Bergantz, G.W. Rhyolites and their Source Mushes across Tectonic Settings. J. Petrol. 2009, 49, 2277–2285. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Chassefière, E.; Leblanc, F.; Brain, D.A. Atmosphere escape and climate evolution of terrestrial planets. In Comparative Climatology of Terrestrial Planets; University of Arizona Press: Tucson, AZ, USA, 2013; pp. 567–581. [Google Scholar]
- Gaillard, F.; Scaillet, B. A theoretical framework for volcanic degassing chemistry in a comparative planetology perspective and implications for planetary atmospheres. Earth Planet. Sci. Lett. 2014, 403, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Kelley, K.A.; Plank, T.; Newman, S.; Stolper, E.M.; Grove, T.L.; Parman, S.; Hauri, E.H. Mantle melting as a function of water content beneath the Mariana Arc. J. Petrol. 2010, 51, 1711–1738. [Google Scholar] [CrossRef] [Green Version]
- Eggler, D.H. Water-saturated and undersaturated melting relations in a Paricutin andesite and an estimate of water content in the natural magma. Contrib. Mineral. Petrol. 1972, 34, 261–271. [Google Scholar] [CrossRef]
- Baker, M.B.; Grove, T.L.; Price, R. Primitive basalts and andesites from the Mt. Shasta region, N. California: Products of varying melt fraction and water content. Contrib. Mineral. Petrol. 1994, 118, 111–129. [Google Scholar] [CrossRef]
- Jamtveit, B.; Brooker, R.; Brooks, K.; Larsen, L.M.; Pedersen, T. The water content of olivines from the North Atlantic Volcanic Province. Earth Planet. Sci. Lett. 2001, 186, 401–415. [Google Scholar] [CrossRef]
- Moore, J.G. Water Content of Basalt Erupted on the ocean floor. Contrib. Mineral. Petrol. 1970, 28, 272–279. [Google Scholar] [CrossRef]
- Geng, J.; Zhang, R.; Wang, X. Chemical Origin of Phosphine in Nature. In Proceedings of the 2010 4th International Conference on Bioinformatics and Biomedical Engineering, Chengdu, China, 18–20 June 2010; pp. 1–12. [Google Scholar]
- Cardelino, B.; Moore, C.; Cardelino, C.; McCall, S.; Frazier, D.; Bachmann, K. Semiclassical calculation of reaction rate constants for homolytical dissociation reactions of interest in organometallic vapor-phase epitaxy (OMVPE). J. Phys. Chem. A 2003, 107, 3708–3718. [Google Scholar] [CrossRef]
- Allison, C.M.; Roggensack, K.; Clarke, A.B. Highly explosive basaltic eruptions driven by CO2 exsolution. Nat. Commun. 2021, 12, 217. [Google Scholar] [CrossRef]
- Krueger, A.J. Sighting of El Chichon sulfur dioxide clouds with the Nimbus 7 total ozone mapping spectrometer. Science 1983, 220, 1377–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, P.K. A comparison of inner Solar System volcanism. Nat. Astron. 2020, 4, 321–327. [Google Scholar] [CrossRef]
- Head, J.W.; Crumpler, L.; Aubele, J.C.; Guest, J.E.; Saunders, R.S. Venus volcanism: Classification of volcanic features and structures, associations, and global distribution from Magellan data. J. Geophys. Res. Planets 1992, 97, 13153–13197. [Google Scholar] [CrossRef] [Green Version]
- Head, J.W., III; Wilson, L. Volcanic processes and landforms on Venus: Theory, predictions, and observations. J. Geophys. Res. Solid Earth 1986, 91, 9407–9446. [Google Scholar] [CrossRef]
- Aramaki, S.; Hayakawa, Y.; Fujii, T.; Nakamura, K.; Fukuoka, T. The October 1983 eruption of Miyakejima volcano. J. Volcanol. Geotherm. Res. 1986, 29, 203–229. [Google Scholar] [CrossRef]
- Parfitt, E.A.; Wilson, L. A Plinian treatment of fallout from Hawaiian lava fountains. J. Volcanol. Geotherm. Res. 1999, 88, 67–75. [Google Scholar] [CrossRef]
- Mastin, L.G.; Guffanti, M.; Servranckx, R.; Webley, P.; Barsotti, S.; Dean, K.; Durant, A.; Ewert, J.W.; Neri, A.; Rose, W.I.; et al. A multidisciplinary effort to assign realistic source parameters to models of volcanic ash-cloud transport and dispersion during eruptions. J. Volcanol. Geotherm. Res. 2009, 186, 10–21. [Google Scholar] [CrossRef]
- Earthchem Collaboration. Earthchem. 2021, Volume 2021. Available online: https://www.earthchem.org/ (accessed on 8 August 2021).
- Sen, G. Generation of Deccan trap magmas. J. Earth Syst. Sci. 2001, 110, 409–431. [Google Scholar] [CrossRef] [Green Version]
- Renne, P.R.; Basu, A.R. Rapid eruption of the Siberian traps flood basalts at the permo-triassic boundary. Science 1991, 253, 176–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasius, K.R.; Cutts, J.A. Topography of Martian central volcanoes. Icarus 1981, 45, 87–112. [Google Scholar] [CrossRef]
- Hardee, H.C.; Larson, D.W. The extraction of heat from magmas based on heat transfer mechanisms. J. Volcanol. Geotherm. Res. 1977, 2, 113–144. [Google Scholar] [CrossRef]
- Sachs, P.M.; Stange, S. Fast assimilation of xenoliths in magmas. J. Geophys. Res. Solid Earth 1993, 98, 19741–19754. [Google Scholar] [CrossRef]
- Robinson, J.E.; Eakins, B.W. Calculated volumes of individual shield volcanoes at the young end of the Hawaiian Ridge. J. Volcanol. Geotherm. Res. 2006, 151, 309–317. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Element | Solid | A (cm2/Day) | Ea (kJ/mol) | Notes | Reference |
|---|---|---|---|---|---|
| P | Iron | 9.84·104 | 306 | Calculated from data in paper | [29] |
| O | Iron | 2.396·10−1 | 118 | Minimally affected by dissolved Si, Al or Mg | [30] |
| Factor | Comment | Section | Multiplier |
|---|---|---|---|
| Phosphine lifetime | could be as specified by Truong and Lunine or full photochemical model | 3.6 | 1–26 |
| Abundance of erupted phosphide, assuming QIF f(O2) | <10−5 of phosphorus likely to be phosphide within 100 km of surface | 3.1, 3.2 | >6000 |
| Delivery of phosphide to clouds | Effusive volcanism at least 10-fold less efficient at producing fine ash as explosive volcanism | 3.3, 3.4 | 101–102 |
| Abundance of phosphorus | Most likely 0.08%, not 1% | 3.5, [11] | 12 |
| Overall scale-up of [16] estimate required to meet PH3 production requirement | ≥7.2·105 | ||
| Volume of volcanic eruption required, based on Truong and Lunine base case of 0.03–0.15 km3/year | ≥26,100 km3/year | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bains, W.; Shorttle, O.; Ranjan, S.; Rimmer, P.B.; Petkowski, J.J.; Greaves, J.S.; Seager, S. Constraints on the Production of Phosphine by Venusian Volcanoes. Universe 2022, 8, 54. https://doi.org/10.3390/universe8010054
Bains W, Shorttle O, Ranjan S, Rimmer PB, Petkowski JJ, Greaves JS, Seager S. Constraints on the Production of Phosphine by Venusian Volcanoes. Universe. 2022; 8(1):54. https://doi.org/10.3390/universe8010054
Chicago/Turabian StyleBains, William, Oliver Shorttle, Sukrit Ranjan, Paul B. Rimmer, Janusz J. Petkowski, Jane S. Greaves, and Sara Seager. 2022. "Constraints on the Production of Phosphine by Venusian Volcanoes" Universe 8, no. 1: 54. https://doi.org/10.3390/universe8010054
APA StyleBains, W., Shorttle, O., Ranjan, S., Rimmer, P. B., Petkowski, J. J., Greaves, J. S., & Seager, S. (2022). Constraints on the Production of Phosphine by Venusian Volcanoes. Universe, 8(1), 54. https://doi.org/10.3390/universe8010054