


GRASP: The Future?


Abstract
:1. Introduction
2. Building Atomic Structure Programs
3. QED of Atoms and Molecules
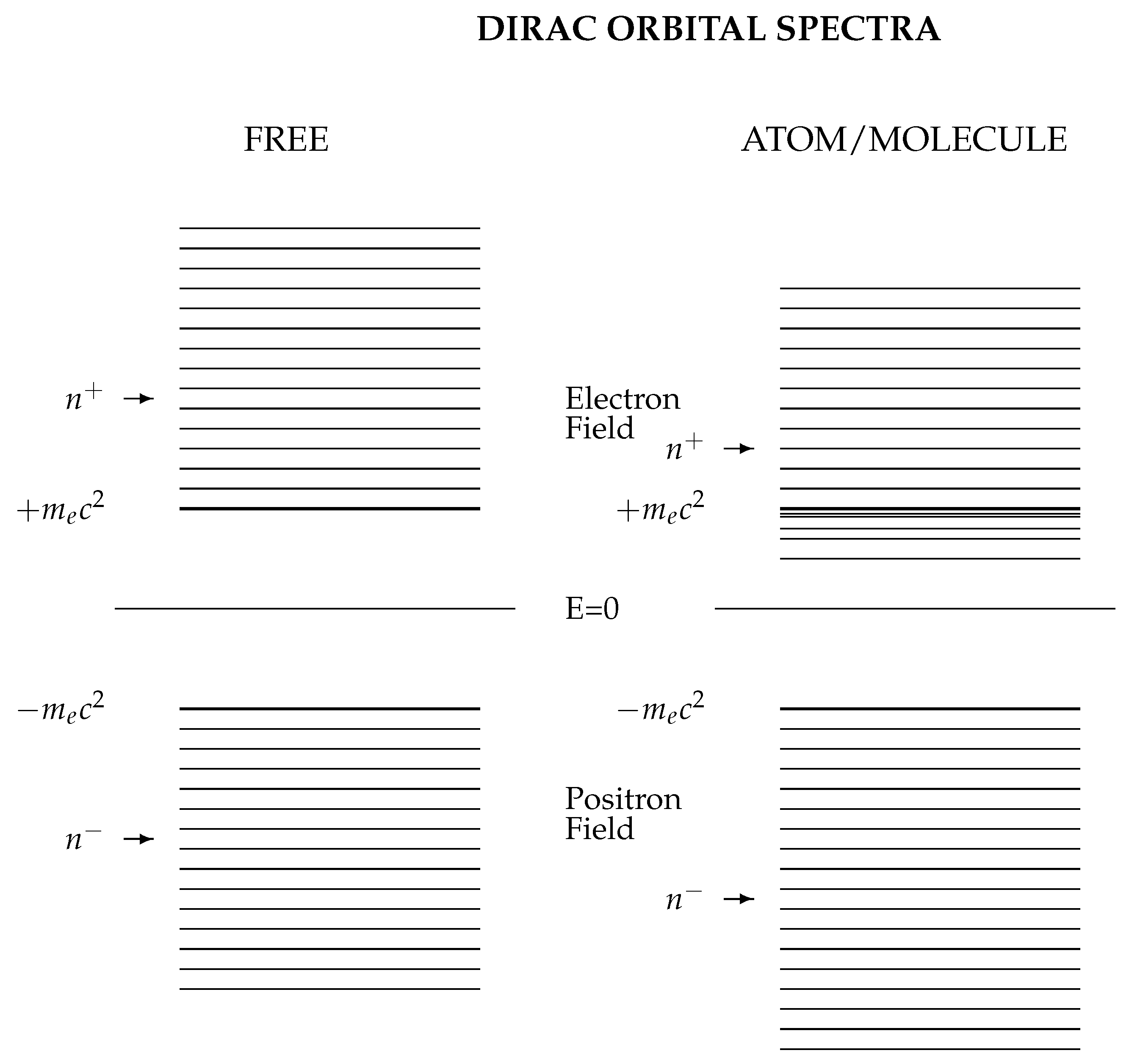
3.1. Relativistic Wave Equations
3.2. Quantized Electron and Positron Fields
3.3. Basis Set Spinor Expansions
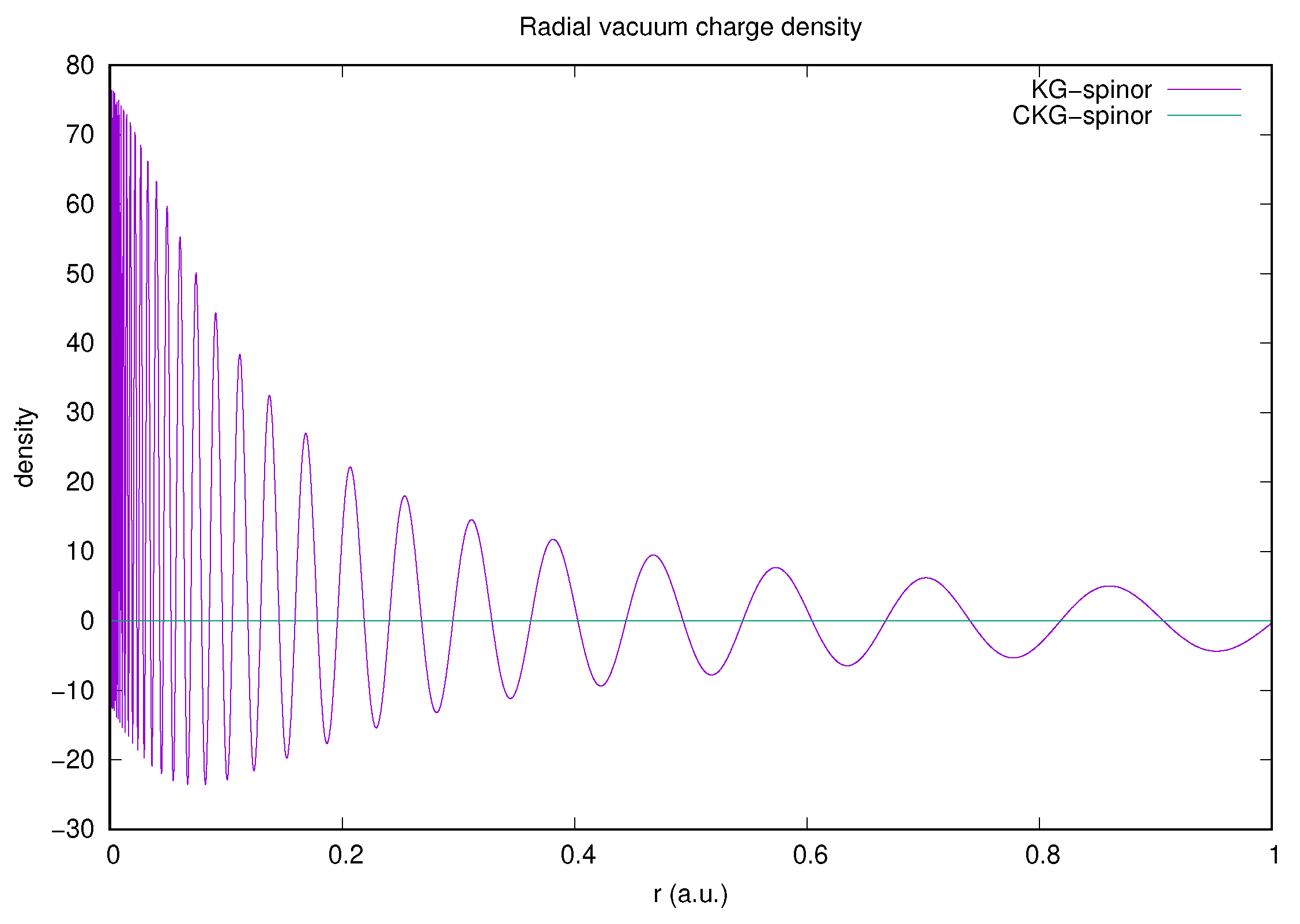
3.3.1. KG-Spinors
3.3.2. Charge Conjugation
3.3.3. CKG-Spinors
4. QED Corrections
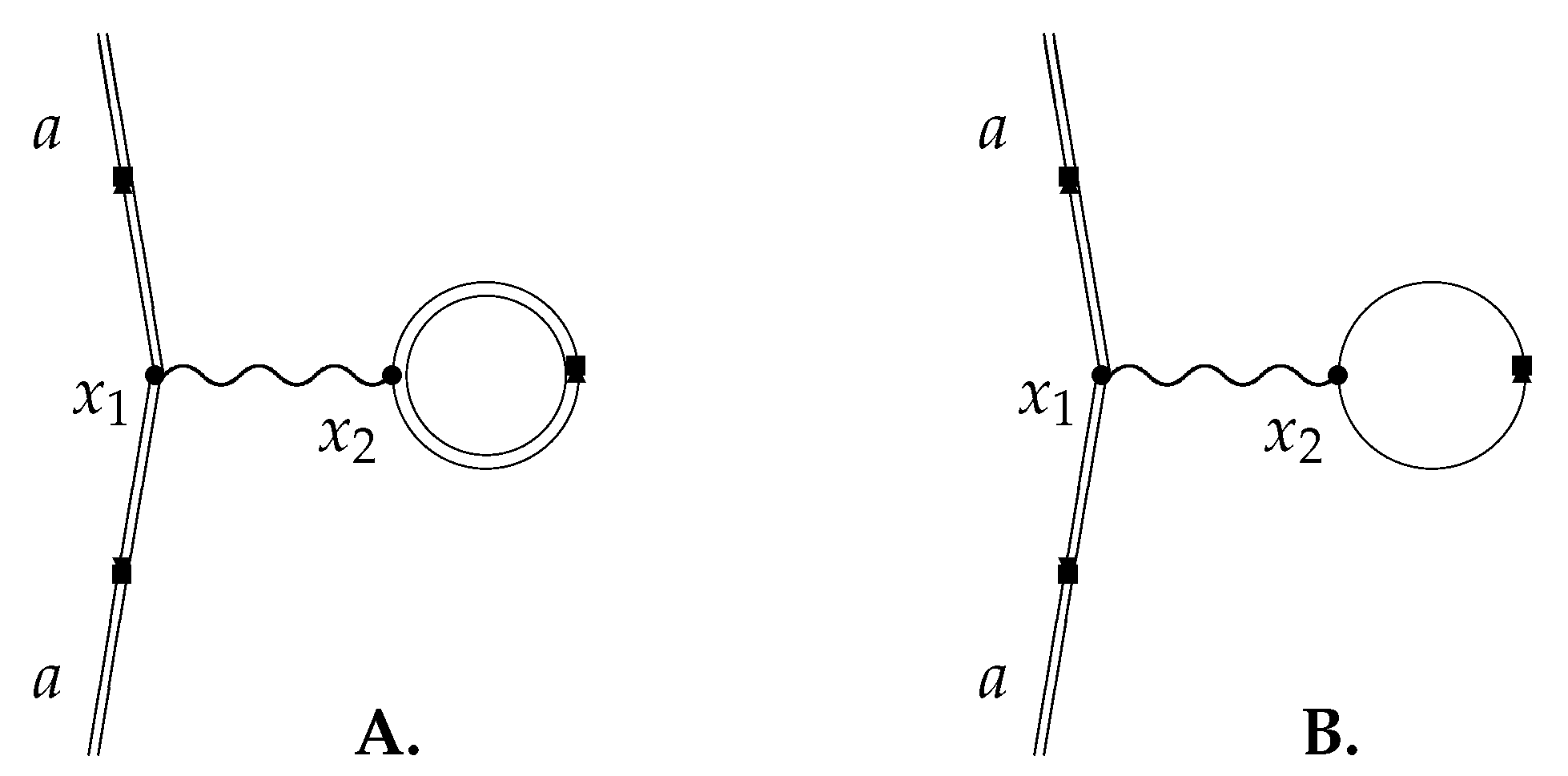
Vacuum Polarization
5. Electron Self-Energy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartree, D.R. The Calculation of Atomic Structures; John Wiley & Sons, Inc.: New York, NY, USA, 1957. [Google Scholar]
- Grant, I.P. Relativistic self-consistent fields. Proc. R. Soc. A 1961, 262, 555–576. [Google Scholar] [CrossRef]
- Grant, I.P. Relativistic calculation of atomic structures. Adv. Phys. 1970, 19, 747. [Google Scholar] [CrossRef]
- Froese Fischer, C.; Gaigalas, G.; Jönsson, P.; Bieroń, J. GRASP2018-A Fortran 95 version of the General Purpose Relativistic Atomic Structure Package. Computer Phys. Commun. 2018, 237, 183–187. [Google Scholar]
- Available online: https://github.com/compas (accessed on 1 June 2022).
- Quiney, H.M.; Skaane, H.; Grant, I.P. Relativistic calculation of electromagnetic interactions in molecules. J. Phys. B At. Mol. Opt. Phys. 1997, 30, L829. [Google Scholar] [CrossRef]
- Belpassi, L.; de Santis, M.; Quiney, H.M.; Tarantelli, F.; Storchi, L. BERTHA: Implementation of a four-component Dirac-Kohn-Sham relativistic framework. J. Chem. Phys. 2020, 152, 164118. [Google Scholar] [CrossRef]
- Grant, I.P. Relativistic Quantum Theory of Atoms and Molecules: Theory and Computation; Springer Science and Business Media, LLC: New York, NY, USA, 2007. [Google Scholar]
- Furry, W.H. On bound states and scattering in positron theory. Phys. Rev. 1951, 81, 115. [Google Scholar] [CrossRef]
- Desclaux, J.-P. Relativistic Dirac-Fock expectation values for atoms Z = 1 to Z = 120. At. Data Nucl. Data Tables 1973, 12, 311–406. [Google Scholar] [CrossRef]
- Johnson, W.R.; Sapirstein, J. Computation of second-order many-body corrections in relativistic atomic systems. Phys. Rev. Lett. 1986, 57, 1126. [Google Scholar] [CrossRef]
- Shabaev, V.M.; Tupitsyn, I.I.; Yerokhin, V.A.; Plunien, G.; Soff, G. Dual kinetic balance approach to basis set expansions for the Dirac equation. Phys. Rev. Lett. 2004, 93, 130405. [Google Scholar] [CrossRef] [Green Version]
- Dyall, K.G.; Faegri, K. Introduction to Relativistic Quantum Chemistry; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Zatsarinny, O.; Froese-Fischer, C. DBSR_HF: A B-spline Dirac-Hartree-Fock program. Comput. Phys. Commun. 2016, 202, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Liu, W. Essentials of relativistic quantum chemistry. J. Chem. Phys. 2020, 152, 180901. [Google Scholar] [CrossRef] [PubMed]
- Saue, T.; Bast, R.; Perero Gomes, A.S.; Jensen, H.A.; Visscher, L.; Aucar, I.A.; di Remigio, R.; Dyall, K.G.; Eliav, E.; Fasshauer, E.; et al. The DIRAC code for relativistic molecular calculations. J. Chem. Phys. 2020, 152, 204104. [Google Scholar] [CrossRef] [PubMed]
- Indelicato, P.; Mohr, P.J.; Sapirstein, J. Coordinate-space approach to vacuum polarization. Phys. Rev. A 2014, 89, 042121. [Google Scholar] [CrossRef] [Green Version]
- Quiney, H.M.; Grant, I.P. Atomic self-energy calculations using partial-wave mass renormalization. J. Phys. B At. Mol. Opt. Phys. 1994, 27, L299–L304. [Google Scholar] [CrossRef]
- Quiney, H.M.; Grant, I.P. Partial-wave mass renormalization in atomic QED calculations. Phys. Scr. 1993, T46, 132–138. [Google Scholar] [CrossRef]
- Grant, I.P.; Quiney, H.M. A class of Bessel function integrals with application in particle physics. J. Phys. A Math. Gen. 1993, 26, 7547–7562. [Google Scholar] [CrossRef]
- Persson, H.; Lindgren, I.; Salomonson, S.; Sunnergren, P. Accurate vacuum-polarization calculations. Phys. Rev. A 1993, 48, 2772–2778. [Google Scholar] [CrossRef]
- Lindgren, I.; Persson, H.; Salomonson, S.; Sunnergren, P. Analysis of the electron self-energy for tightly bound electrons. Phys. Rev. A 1998, 58, 1001–1015. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://compas.github.io (accessed on 1 June 2022).
- Roothaan, C.C.J. New developments in molecular orbital theory. Rev. Mod. Phys. 1951, 23, 69. [Google Scholar] [CrossRef]
- Hall, G.G. The molecular orbital theory of chemical valency VIII: A method of calculating ionization potentials. Proc. R. Soc. A 1951, 205, 541. [Google Scholar]
- Pais, A. Inward Bound; Oxford University Press: Oxford UK, 1986. [Google Scholar]
- Schweber, S.S. QED and the Men Who Made It; Princeton University Press: Princeton, NJ, USA, 1994. [Google Scholar]
- Weinberg, S. Quantum Theory of Fields, Vol 1; Cambridge University Press: Cambridge UK, 1995. [Google Scholar]
- Dirac, P.A.M. The quantum theory of the electron, I. Proc. R. Soc. A 1928, 117, 610. [Google Scholar]
- Dirac, P.A.M. The quantum theory of the electron, II. Proc. R. Soc. A 1928, 118, 351. [Google Scholar]
- Dirac, P.A.M. A theory of electrons and protons. Proc. R. Soc. A 1930, 126, 360. [Google Scholar]
- Gordon, W. Der Strom der Diracschen Elektrontheorie. Z. Phys. 1928, 48, 11. [Google Scholar] [CrossRef]
- Darwin, C.G. The wave equations of the electron. Proc. R. Soc. A 1928, 118, 654. [Google Scholar]
- Wichmann, E.H.; Kroll, N.M. Vacuum polarization in a strong Coulomb field. Phys. Rev. 1956, 101, 843. [Google Scholar] [CrossRef]
- Blomqvist, J. Vacuum polarization in exotic atoms. Nucl. Phys. B 1972, 48, 95. [Google Scholar] [CrossRef]
- Furry, W.H. A symmetry theorem in the positron theory. Phys. Rev. 1937, 51, 125. [Google Scholar] [CrossRef]
- Schweber, S.S. Relativistic Quantum Field Theory; Harper and Row: New York, NY, USA, 1961. [Google Scholar]
- Sapirstein, J.; Cheng, K.T. Vacuum polarization calculations for hydrogenlike and alkali-metal-like ions. Phys. Rev. 2003, 68, 042111. [Google Scholar] [CrossRef] [Green Version]
- Feynman, R.P. The theory of positrons. Phys. Rev. 1949, 76, 749. [Google Scholar] [CrossRef]
- Feynman, R.P. Space-time approach to quantum electrodynamics. Phys. Rev. 1949, 76, 769. [Google Scholar] [CrossRef] [Green Version]
- Racah, G. Theory of complex spectra: II. Phys. Rev. 1942, 62, 438. [Google Scholar] [CrossRef]
- Racah, G. Theory of complex spectra: III. Phys. Rev. 1943, 63, 367. [Google Scholar] [CrossRef]
- Racah, G. Theory of complex spectra: IV. Phys. Rev. 1949, 76, 1352. [Google Scholar] [CrossRef]
- Salman, M.; Saue, T. Charge conjugation symmetry in the finite basis approximation of the Dirac equation. Symmetry 2020, 12, 1121. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| −1 | −3532.1921489294 | −3532.1921489289 | −904.8478012876 | −392.0836928780 |
| 1 | −904.8478012882 | −904.8478012878 | −392.0836928781 | −216.4247478039 |
| −2 | −817.8074977480 | −817.8074977480 | −366.1427114567 | −205.5771277760 |
| 2 | −366.1427114567 | −366.1427114567 | −205.5771277760 | −131.1010555098 |
| −3 | −358.9868485160 | −358.9868485160 | −202.5363034958 | −129.6328330777 |
| 3 | −202.5363034958 | −202.5363034958 | −129.6328330776 | −89.9621990162 |
| −4 | −201.0765233582 | −201.0765233582 | −128.8823613985 | −89.5273365309 |
| 4 | −128.8823613985 | −128.8823613985 | −89.5273365309 | −65.7655876909 |
| −5 | −128.4392341889 | −128.4392341889 | −89.2702733629 | −65.6035374887 |
| 5 | −89.2702733629 | −89.2702733629 | −65.6035374887 | −50.2279904000 |
| −6 | −89.1002663743 | −89.1002663743 | −65.4963124418 | −50.1561023779 |
| 6 | −65.4963124418 | −65.4963124419 | −50.1561023780 | −39.6314776684 |
| −7 | −65.4200746697 | −65.4200746697 | −50.1049761176 | −39.5955492450 |
| 7 | −50.1049761176 | −50.1049761176 | −39.5955492450 | −32.0742810655 |
| −8 | −50.0667420260 | −50.0667420260 | −39.5686766900 | −32.0546823612 |
| 8 | −39.5686766900 | −39.5686766900 | −32.0546823613 | −26.4929690886 |
| −9 | −39.5478161969 | −39.5478161969 | −32.0394670086 | −26.4815336906 |
| 9 | −32.0394670087 | −32.0394670087 | −26.4815336906 | −22.2529707815 |
| −10 | −32.0273112566 | −32.0273112566 | −26.4723972662 | −22.2459315332 |
| 10 | −26.4723972662 | −26.4723972662 | −22.2459315332 | −18.9559506743 |
| 1 | 3.29166528 | 3.22434547 | 6.73198 (−2) |
| 2 | 2.81964973 | 2.81325408 | 6.39565 (−3) |
| 3 | 2.40679616 | 2.40522784 | 1.56831 (−3) |
| 4 | 2.07156051 | 2.07095146 | 6.09053 (−4) |
| 5 | 1.79881093 | 1.79850086 | 3.10045 (−4) |
| 6 | 1.57229887 | 1.57211452 | 1.84348 (−4) |
| 7 | 1.38083155 | 1.38071232 | 1.29222 (−4) |
| 8 | 1.21719228 | 1.21711175 | 8.05279 (−5) |
| Sum | 16.5588053 | 16.42822183 | 7.65869 (−2) |
| Z | |||
|---|---|---|---|
| 10 | 3.232201 (−7) | 3.740969 (−7) | 3.714822 (−7) |
| 20 | 1.862744 (−5) | 2.036149 (−5) | 2.042291 (−5) |
| 30 | 1.964703 (−4) | 2.093781 (−4) | 2.114400 (−4) |
| 40 | 1.046683 (−3) | 1.098815 (−3) | 1.120773 (−3) |
| 50 | 3.869888 (−3) | 4.023198 (−3) | 4.158461 (−3) |
| 60 | 1.144670 (−2) | 1.181736 (−2) | 1.242842 (−2) |
| 70 | 2.926072 (−2) | 3.005133 (−2) | 3.230899 (−2) |
| 80 | 6.784298 (−2) | 6.935930 (−2) | 7.667064 (−2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grant, I.; Quiney, H. GRASP: The Future? Atoms 2022, 10, 108. https://doi.org/10.3390/atoms10040108
Grant I, Quiney H. GRASP: The Future? Atoms. 2022; 10(4):108. https://doi.org/10.3390/atoms10040108
Chicago/Turabian StyleGrant, Ian, and Harry Quiney. 2022. "GRASP: The Future?" Atoms 10, no. 4: 108. https://doi.org/10.3390/atoms10040108
APA StyleGrant, I., & Quiney, H. (2022). GRASP: The Future? Atoms, 10(4), 108. https://doi.org/10.3390/atoms10040108