
DFT Investigation on the Complexation of β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin as Recognition Hosts with Trichloroethylene

 , , ,
, , ,
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
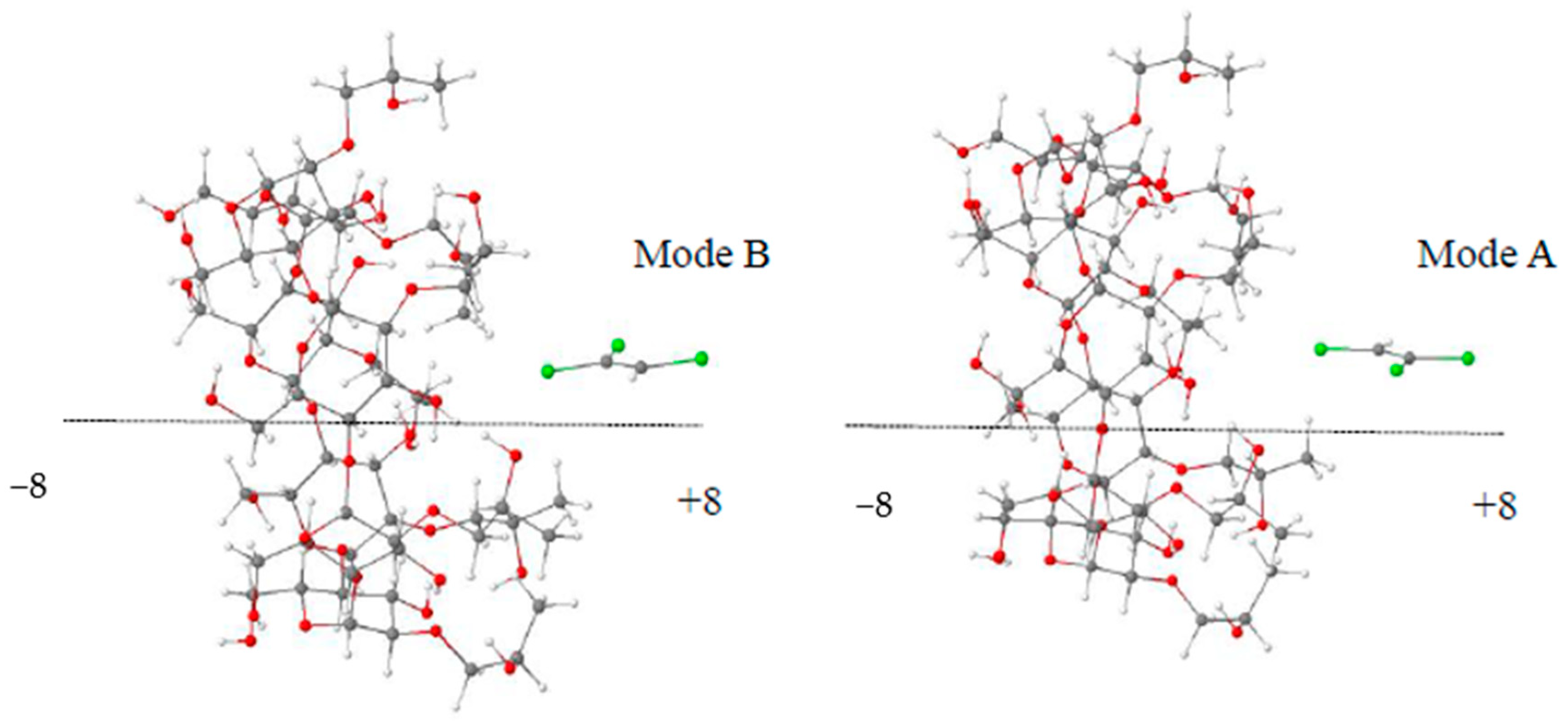
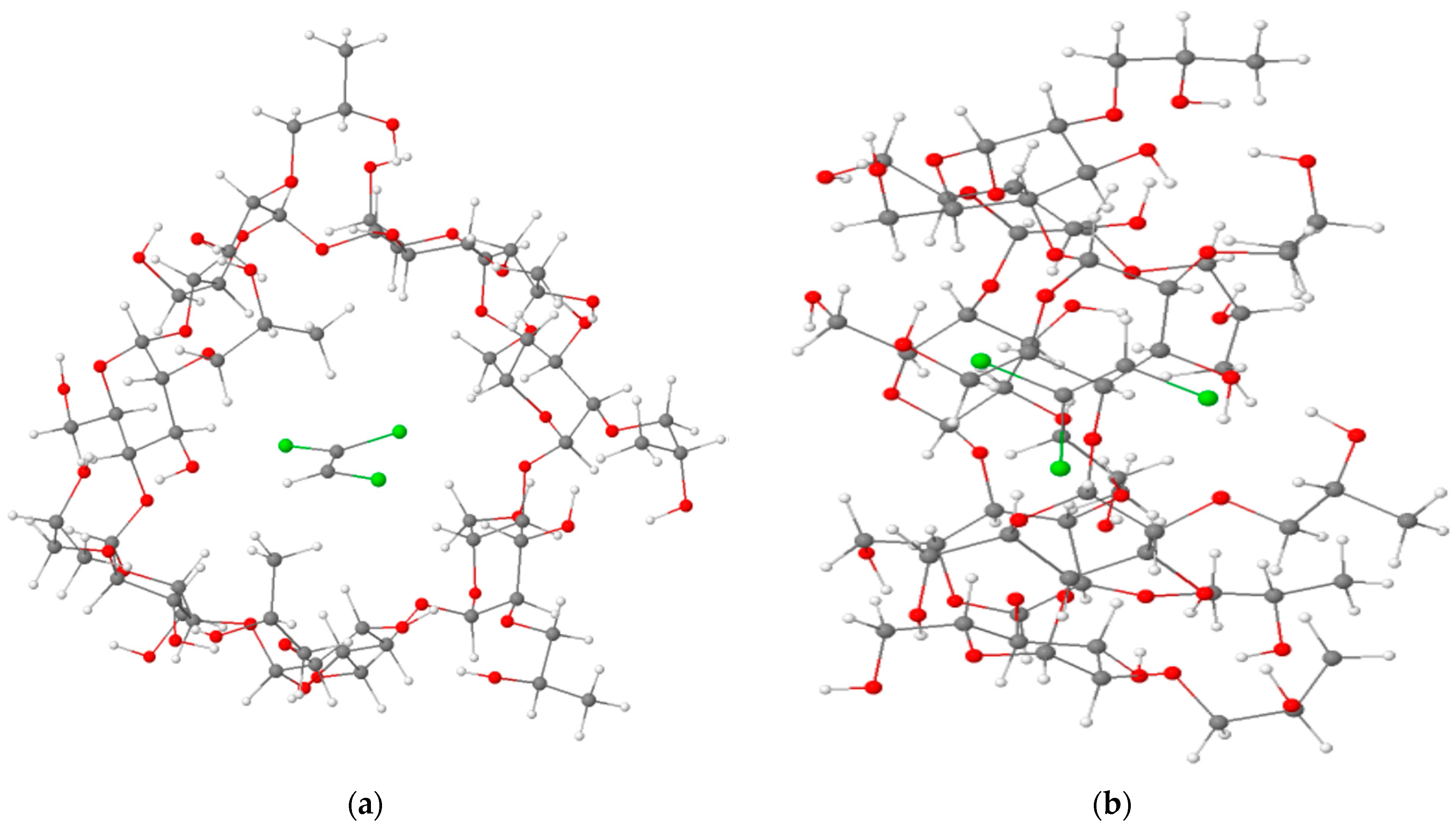
3.1. Calculations of Complexation Energies
3.2. Thermodynamics Properties of the Inclusion Processes
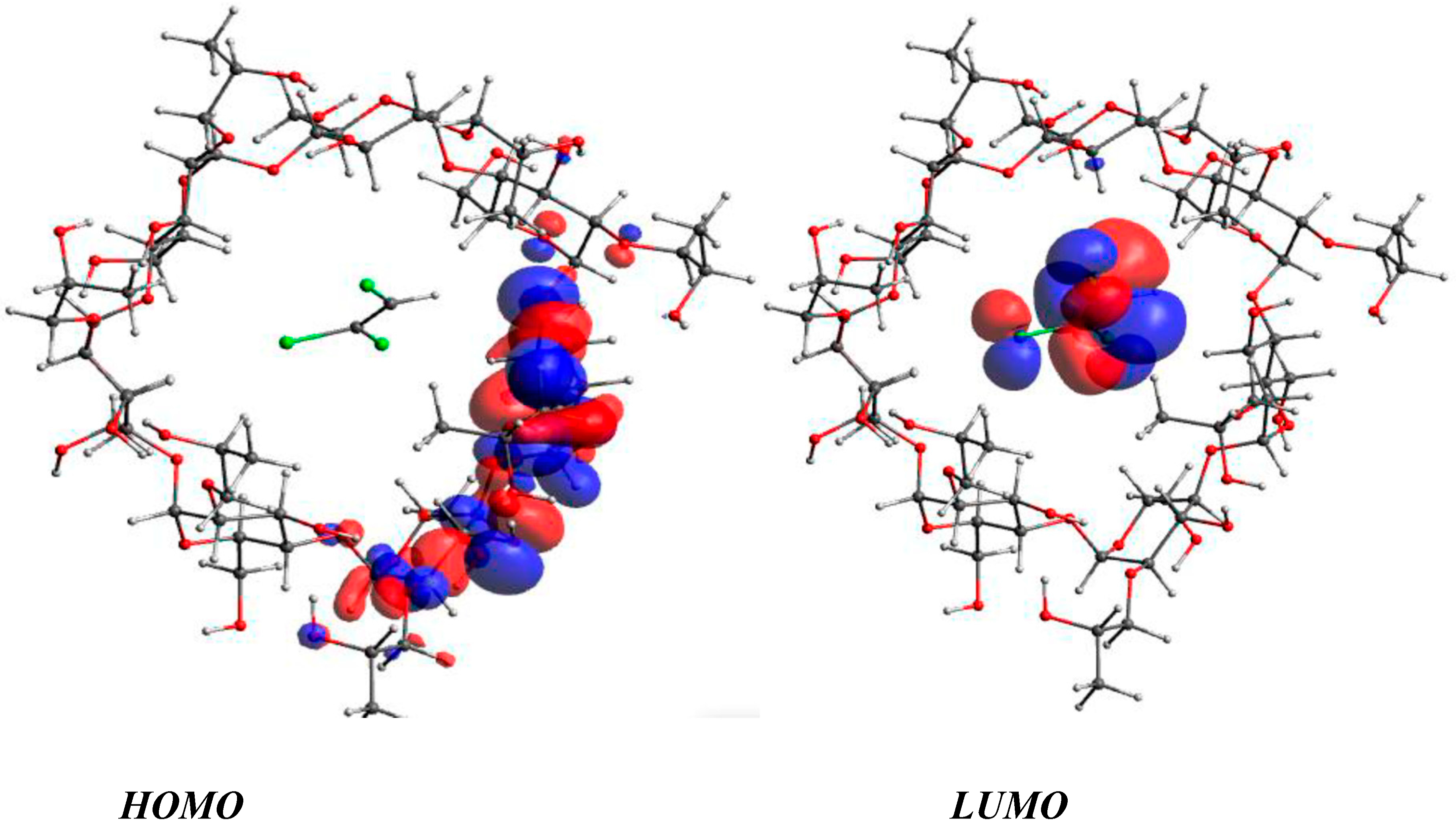
3.3. DFT Calculations of HOMO, LUMO, HOMO–LUMO Energy Gap, and Dipole Moment
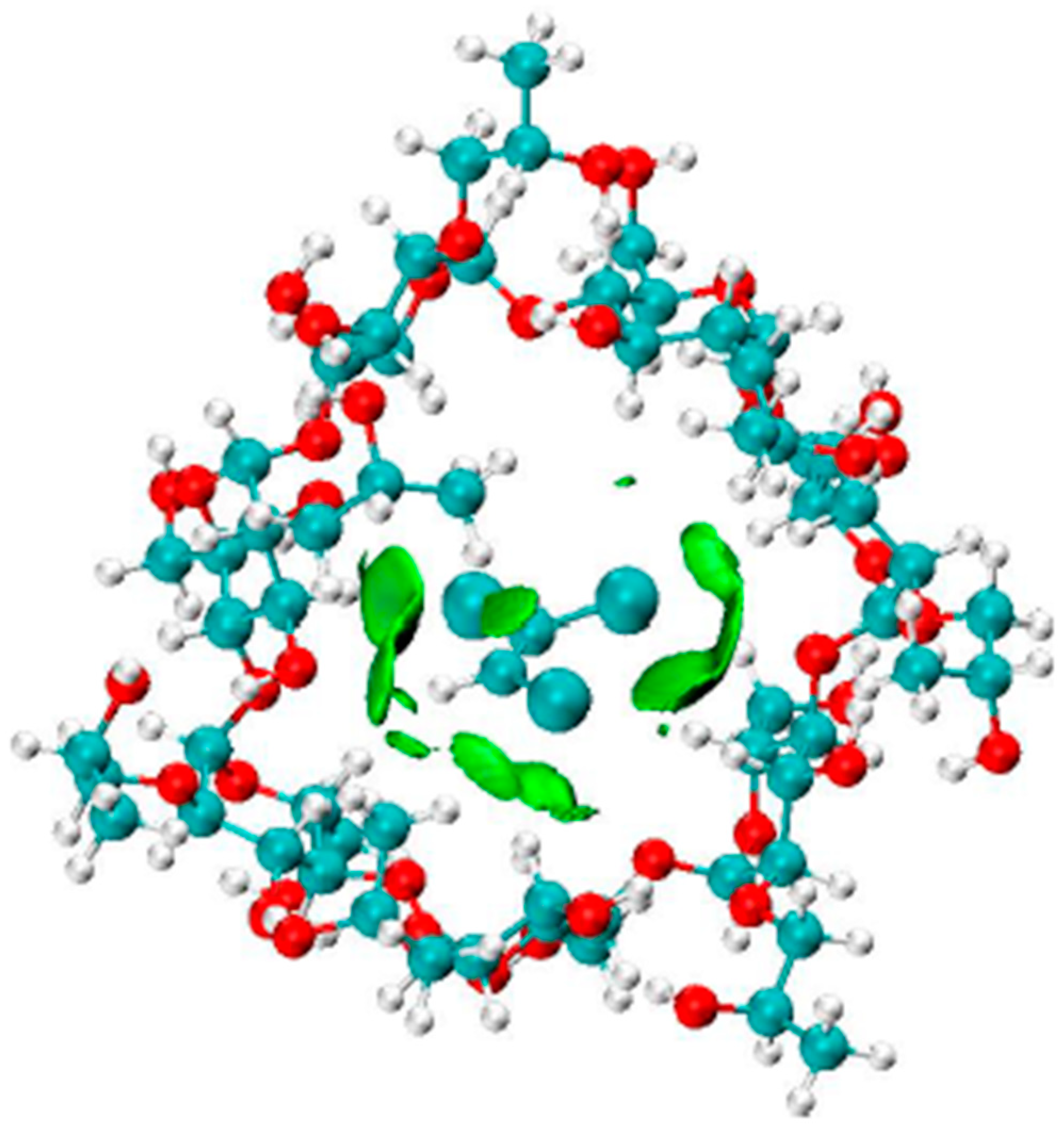
3.4. Characterization of the Non-Covalent Intermolecular Interactions
3.5. Charge Decomposition Analysis
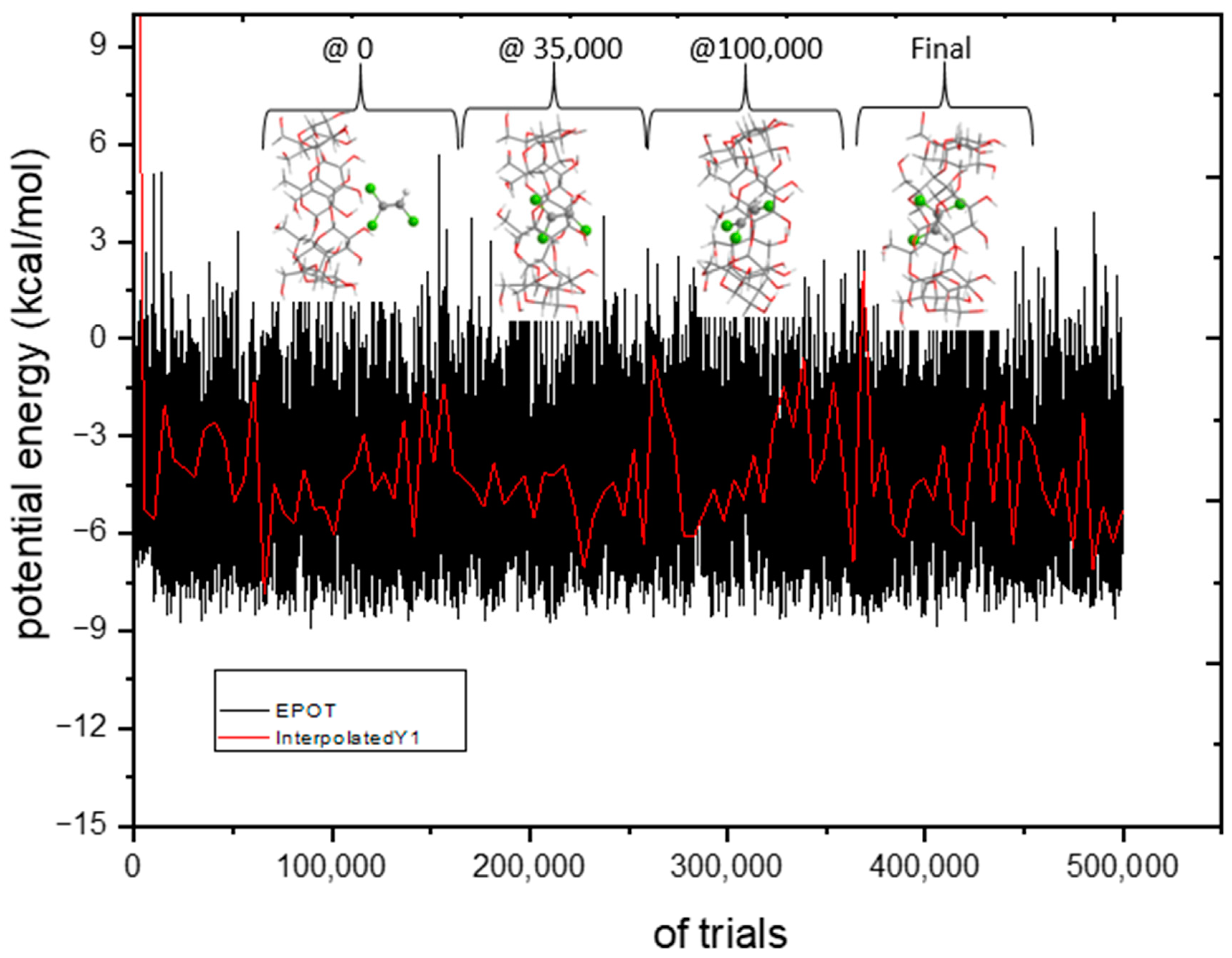
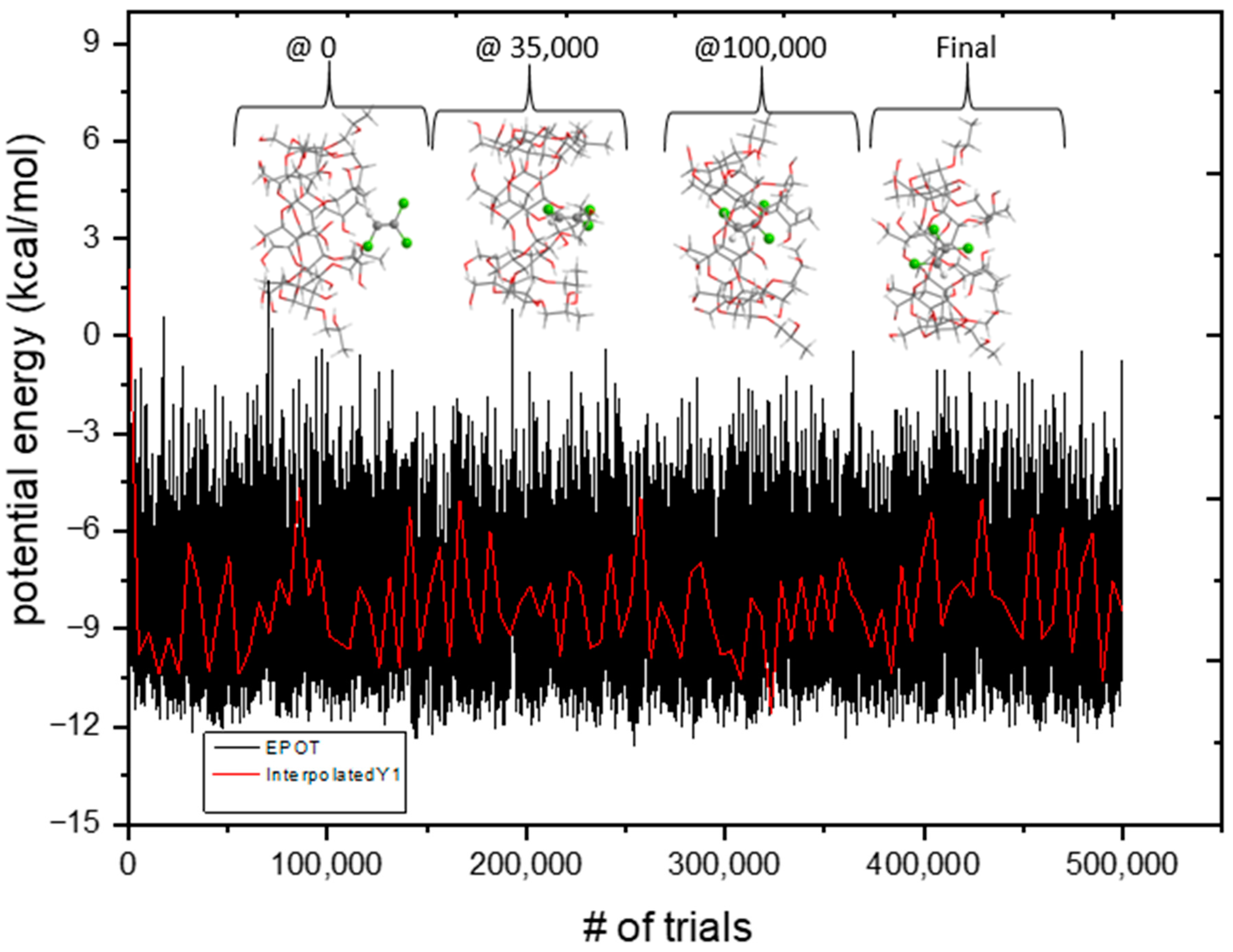
3.6. Monte Carlo Docking Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Antoine, R. Supramolecular Gold Chemistry: From Atomically Precise Thiolate-Protected Gold Nanoclusters to Gold-Thiolate Nanostructures. J. Nanomater. 2020, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, G. Analytical supramolecular chemistry: Colorimetric and fluorimetric chemosensors. J. Photochem. Photobiol. C Photochem. Rev. 2020, 42, 100340. [Google Scholar] [CrossRef]
- Lehn, J.-M. From supramolecular chemistry towards constitutional dynamic chemistry and adaptive chemistry. Chem. Soc. Rev. 2007, 36, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.-H.; Luo, J.; Mao, Y.-L.; Lai, S.; Gong, Y.-N.; Zhong, D.-C.; Lu, T.-B. π-π stacking interactions: Non-negligible forces for stabilizing porous supramolecular frameworks. Sci. Adv. 2020, 6, eaax9976. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.J. The discovery of crown ethers. Science 1988, 241, 536–540. [Google Scholar] [CrossRef]
- Freeman, W.A.; Mock, W.L.; Shih, N.-Y. Cucurbituril. J. Am. Chem. Soc. 1981, 103, 7367–7368. [Google Scholar] [CrossRef]
- Del Valle, E.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Gutsche, C.D. Calixarenes. Acc. Chem. Res. 1983, 16, 161–170. [Google Scholar] [CrossRef]
- Yadav, M.; Thakore, S.; Jadeja, R. A review on remediation technologies using functionalized Cyclodextrin. Environ. Sci. Pollut. Res. 2022, 29, 236–250. [Google Scholar] [CrossRef]
- Gonzalez Pereira, A.; Carpena, M.; García Oliveira, P.; Mejuto, J.C.; Prieto, M.A.; Simal Gandara, J. Main applications of cyclodextrins in the food industry as the compounds of choice to form host–guest complexes. Int. J. Mol. Sci. 2021, 22, 1339. [Google Scholar] [CrossRef]
- Aiassa, V.; Garnero, C.; Longhi, M.R.; Zoppi, A. Cyclodextrin multicomponent complexes: Pharmaceutical applications. Pharmaceutics 2021, 13, 1099. [Google Scholar] [CrossRef] [PubMed]
- Rincón-López, J.; Almanza-Arjona, Y.C.; Riascos, A.P.; Rojas-Aguirre, Y. Technological evolution of cyclodextrins in the pharmaceutical field. J. Drug Deliv. Sci. Technol. 2021, 61, 102156. [Google Scholar] [CrossRef] [PubMed]
- Crini, G.; Fenyvesi, É.; Szente, L. Outstanding contribution of Professor József Szejtli to cyclodextrin applications in foods, cosmetics, drugs, chromatography and biotechnology: A review. Environ. Chem. Lett. 2021, 19, 2619–2641. [Google Scholar] [CrossRef]
- Roy, I.; Stoddart, J.F. Cyclodextrin metal–organic frameworks and their applications. Acc. Chem. Res. 2021, 54, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, T.; Cheng, C.; Wang, Q.; Lin, S.; Liu, C.; Han, X. Research progress on synthesis and application of cyclodextrin polymers. Molecules 2021, 26, 1090. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Renedo, J.M.; Hernández-Esparza, R.; Cuevas-Yañez, E.; Reyes-Pérez, H.; Vargas, R.; Garza, J.; González-Rivas, N. Deformations of cyclodextrins and their influence to form inclusion compounds. Int. J. Quantum Chem. 2021, 122, e26859. [Google Scholar] [CrossRef]
- Nelumdeniya, N.R.M.; Ranatunga, R.J.K.U. Complex forming behaviour of α, β and γ-cyclodextrins with varying size probe particles in silico. Ceylon J. Sci. 2021, 50, 329–339. [Google Scholar] [CrossRef]
- Tian, B.; Hua, S.; Tian, Y.; Liu, J. Cyclodextrin-based adsorbents for the removal of pollutants from wastewater: A review. Environ. Sci. Pollut. Res. 2021, 28, 1317–1340. [Google Scholar] [CrossRef]
- Majd, M.; Yazdanpanah, M.; Bayatloo, M.R.; Nojavan, S. Recent advances and applications of cyclodextrins in magnetic solid phase extraction. Talanta 2021, 229, 122296. [Google Scholar] [CrossRef]
- Kim, J.S.; Choi, Y.J.; Woo, M.R.; Cheon, S.; Ji, S.H.; Im, D.; Ud Din, F.; Kim, J.O.; Youn, Y.S.; Oh, K.T.; et al. New potential application of hydroxypropyl-β-cyclodextrin in solid self-nanoemulsifying drug delivery system and solid dispersion. Carbohydr. Polym. 2021, 271, 118433. [Google Scholar] [CrossRef]
- Oba, B.T.; Zheng, X.; Aborisade, M.A.; Liu, J.; Yohannes, A.; Kavwenje, S.; Sun, P.; Yang, Y.; Zhao, L. Remediation of trichloroethylene contaminated soil by unactivated peroxymonosulfate: Implication on selected soil characteristics. J. Environ. Manag. 2021, 285, 112063. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, H.; Zhao, L.; Li, Z.; Yi, X.; Guo, T.; Cao, X. Enhanced trichloroethylene biodegradation: Roles of biochar-microbial collaboration beyond adsorption. Sci. Total Environ. 2021, 792, 148451. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xia, Y.; Tao, Y.; Jin, H.; Ji, C.; Aniagu, S.; Chen, T.; Jiang, Y. Protective effects of resveratrol against the cardiac developmental toxicity of trichloroethylene in zebrafish embryos. Toxicology 2021, 452, 152697. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry (ATSDR). ATSDR’s Substance Priority List. 2017. Available online: https://www.atsdr.cdc.gov/spl/resources/2017_atsdr_substance_priority_list.html (accessed on 19 July 2022).
- Love, O.T., Jr.; Eilers, R.G. Treatment of drinking water containing trichloroethylene and related industrial solvents. J.-Am. Water Works Assoc. 1982, 74, 413–425. [Google Scholar] [CrossRef]
- Hennebel, T.; Simoen, H.; De Windt, W.; Verloo, M.; Boon, N.; Verstraete, W. Biocatalytic dechlorination of trichloroethylene with bio-palladium in a pilot-scale membrane reactor. Biotechnol. Bioeng. 2009, 102, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xu, G.; Tu, Y.; Zhang, W.; Hu, X.; Yang, P.; Xie, X. Multifunctional porous β-cyclodextrin polymer for water purification. Water Res. 2022, 222, 118917. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, T.T.; Peng, H.K.; Ren, H.T.; Lou, C.W.; Lin, J.H. Low-cost hydrogel adsorbent enhanced by trihydroxy melamine and β-cyclodextrin for the removal of Pb (II) and Ni (II) in water. J. Hazard. Mater. 2021, 411, 125029. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, M.; Mu, X.; Zhou, J.; Ke, X.; Wu, Q.; Miao, L. Sustainable β-cyclodextrin modified polyacrylamide hydrogel for highly efficient solar-driven water purification. Mater. Today Energy 2023, 35, 101330. [Google Scholar] [CrossRef]
- Morin-Crini, N.; Crini, G. Environmental applications of water-insoluble β-cyclodextrin–epichlorohydrin polymers. Prog. Polym. Sci. 2013, 38, 344–368. [Google Scholar] [CrossRef]
- Mahmood, A.; Khan, S.U.-D.; Ur Rehman, F. Assessing the quantum mechanical level of theory for prediction of UV/Visible absorption spectra of some aminoazobenzene dyes. J. Saudi Chem. Soc. 2015, 19, 436–441. [Google Scholar] [CrossRef]
- Chermette, H.J. Chemical reactivity indexes in density functional theory. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Belhocine, Y.; Bouhadiba, A.; Rahim, M.; Nouar, L.; Djilani, I.; Khatmi, D.I. Inclusion Complex Formation of β-Cyclodextrin with the Nonsteroidal Anti-inflammatory Drug Flufenamic Acid: Computational Study. Macroheterocycles 2018, 11, 203–209. [Google Scholar] [CrossRef]
- Belhocine, Y.; Rahali, S.; Allal, H.; Assaba, I.M.; Ghoniem, M.G.; Ali, F.A.M. A dispersion corrected DFT investigation of the inclusion complexation of dexamethasone with β-cyclodextrin and molecular docking study of its potential activity against COVID-19. Molecules 2021, 26, 7622. [Google Scholar] [CrossRef] [PubMed]
- Shirin, S.; Buncel, E.; vanLoon, G.W. The use of beta-cyclodextrins to enhance the aqueous solubility of trichloroethylene and perchloroethylene and their removal from soil organic matter: Effect of substituents. Can. J. Chem. 2003, 81, 45–52. [Google Scholar] [CrossRef]
- Kashiyama, N.; Boving, T.B. Hindered gas-phase partitioning of trichloroethylene from aqueous cyclodextrin systems: Implications for treatment and analysis. Environ. Sci. Technol. 2004, 38, 4439–4444. [Google Scholar] [CrossRef]
- Liang, C.; Huang, C.; Mohanty, N.; Lu, C.J.; Kurakalva, R.M. Hydroxypropyl-beta-cyclodextrin-mediated iron-activated persulfate oxidation of trichloroethylene and tetrachloroethylene. Ind. Eng. Chem. Res. 2007, 46, 6466–6479. [Google Scholar] [CrossRef]
- Khan, N.A.; Johnson, M.D.; Carroll, K.C. Spectroscopic Methods for Aqueous Cyclodextrin Inclusion Complex Binding Measurement for 1,4-Dioxane, Chlorinated Co-Contaminants, and Ozone. J. Contam. Hydrol. 2018, 210, 31–41. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Aree, T.; Chaichit, N. Crystal structure of β-cyclodextrin–benzoic acid inclusion complex. Carbohydr. Res. 2003, 338, 439–446. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef] [PubMed]
- Kruse, H.; Grimme, S.A. geometrical correction for the inter-and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Guo, Q.X. Use of quantum chemical methods to study cyclodextrin chemistry. J. Incl. Phenom. Macrocycl. Chem. 2004, 50, 95–103. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org (accessed on 3 August 2022).
- Dapprich, S.; Frenking, G. Investigation of donor-acceptor interactions: A charge decomposition analysis using fragment molecular orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Xiao, M.; Lu, T. Generalized Charge Decomposition Analysis (GCDA) Method. J. Adv. Phys. Chem. 2015, 4, 111–124. [Google Scholar] [CrossRef]
- Gorelsky, S.I.; Ghosh, S.; Solomon, E.I. Mechanism of N2O reduction by the μ4-S tetranuclear CuZ cluster of nitrous oxide reductase. J. Am. Chem. Soc. 2006, 128, 278–290. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, R.G. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q. Independent Gradient Model Based on Hirshfeld Partition: A New Method for Visual Study of Interactions in Chemical Systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Mesri, N.; Belhocine, Y.; Messikh, N.; Sayede, A.; Mouffok, B. Molecular DFT Investigation on the Inclusion Complexation of Benzo [a] pyrene with γ-Cyclodextrin. Macroheterocycles 2021, 14, 164–170. [Google Scholar] [CrossRef]
- Messiad, F.A.; Ammouchi, N.; Belhocine, Y.; Alhussain, H.; Ghoniem, M.G.; Said, R.B.; Ali, F.A.M.; Rahali, S. In Search of Preferential Macrocyclic Hosts for Sulfur Mustard Sensing and Recognition: A Computational Investigation through the New Composite Method r2SCAN-3c of the Key Factors Influencing the Host-Guest Interactions. Nanomaterials 2022, 12, 2517. [Google Scholar] [CrossRef]
- Kabouche, Z.; Belhocine, Y.; Benlecheb, T.; Assaba, I.M.; Litim, A.; Lalalou, R.; Mechhoud, A. A DFT-D4 investigation of the complexation phenomenon between pentachlorophenol and β-cyclodextrin. Chim. Techno Acta 2023, 10, 202310209. [Google Scholar] [CrossRef]
- Litim, A.; Belhocine, Y.; Benlecheb, T.; Ghoniem, M.G.; Kabouche, Z.; Ali, F.A.M.; Abdulkhair, B.Y.; Seydou, M.; Rahali, S. DFT-D4 Insight into the Inclusion of Amphetamine and Methamphetamine in Cucurbituril: Energetic, Structural and Biosensing Properties. Molecules 2021, 26, 7479. [Google Scholar] [CrossRef]
- Assaba, I.M.; Rahali, S.; Belhocine, Y.; Allal, H. Inclusion complexation of chloroquine with α and β-cyclodextrin: Theoretical insights from the new B97-3c composite method. J. Mol. Struct. 2021, 1227, 129696. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Singh, U.C.; Case, D.A.; Ghio, C.; Alagona, G.; Profeta, S.; Weiner, P. A New Force Field for Molecular Mechanical Simulation of Nucleic Acids and Proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Nguyen, D.T.; Case, D.A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Bonnet, P.; Beà, I.; Jaime, C.; Morin-Allory, L. Molecular Modelling Study of the 2:1 γ-Cyclodextrin:C60 Complex. Dummy Atoms Simulating Bond Electron Distribution. Supramol. Chem. 2003, 15, 251–260. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hyperchem, Release. 7.51 for Windows MM System; Hypercube, Inc.: Gainesville, FL, USA, 2002.
- Murthy, C.N.; Geckeler, K.E. Stability studies on the water-soluble β-cyclodextrin–Fullerene inclusion complex. Fuller. Nanotub. Carbon Nanostruct. 2002, 10, 91–98. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Configurations | TCE@β-CD Mode A | TCE@β-CD Mode B | TCE@HP-β-CD Mode A | TCE@HP-β-CD Mode B |
|---|---|---|---|---|
| −8 | −12.21 | −16.42 | −12.51 | −14.04 |
| −6 | −16.73 | −16.49 | −12.50 | −13.55 |
| −4 | −16.73 | −18.44 | −21.20 | −15.78 |
| −2 | −16.73 | −16.80 | −16.53 | −15.78 |
| 0 | −17.77 | −16.78 | −16.54 | −16.28 |
| 2 | −14.42 | −17.30 | −16.43 | −17.12 |
| 4 | −14.44 | −14.20 | −16.63 | −14.87 |
| 6 | −12.38 | −15.72 | −16.72 | −12.77 |
| 8 | −12.82 | −15.49 | −12.95 | −14.71 |
| Energetic Parameters | TCE@HP-β-CD (−4A) | TCE@β-CD (−4B) |
|---|---|---|
| ΔEcomplexation (kcal/mol) | −21.20 | −18.44 |
| ΔH° (kcal/mol) | −19.34 | −16.08 |
| ΔG° (kcal/mol) | −5.02 | −1.79 |
| T(ΔS°) (kcal/mol) | −14.31 | −14.29 |
| Parameters | HP-β-CD | β-CD | TCE | TCE@HP-β-CD | TCE@β-CD |
|---|---|---|---|---|---|
| EHOMO (eV) (gas) | −5.18 (−7.88) | −5.24 (−8.06) | −5.78 (−8.15) | −5.37 (−8.07) | −5.32 (−8.18) |
| EHOMO (eV) (water) | −5.27 (−8.07) | −5.55 (−8.34) | −5.72 (−8.09) | −5.26 (−8.07) | −5.57 (−8.21) |
| ELUMO (eV) (gas) | −0.22 (2.24) | −0.59 (1.94) | −1.39 (−0.02) | −1.58 (−0.14) | −2.07 (−0.63) |
| ELUMO (eV) (water) | 0.43 (2.44) | 0.50 (2.75) | −1.27 (0.10) | −1.48 (−0.02) | −1.49 (−0.07) |
| ΔEGap (eV) (gas) | 4.96 (10.12) | 4.65 (10.00) | 4.39 (8.13) | 3.79 (7.93) | 3.25 (7.55) |
| ΔEGap (eV) (water) | 5.70 (10.51) | 6.05 (11.09) | 4.45 (8.19) | 3.78 (8.05) | 4.08 (8.14) |
| μ (Debye) (gas) | 6.61 (6.48) | 9.39 (9.86) | 0.71 (0.93) | 7.30 (7.57) | 10.54 (10.79) |
| μ (Debye) (water) | 8.63 (9.95) | 10.84 (8.88) | 1.13 (1.39) | 10.71 (11.80) | 13.35 (13.26) |
| CDA | ECDA | ||||
|---|---|---|---|---|---|
| Complex | d | b | d−b | r | Net Electrons Obtained by Hosts |
| TCE@β-CD | 0.124 | 0.010 | 0.114 | −0.045 | 0.146 |
| TCE@HP-β-CD | 0.117 | 0.031 | 0.085 | −0.050 | 0.111 |
| a〈Ep〉 | ΔEC | Eint | Edef (TCE) | Edef (β-CD) | Edef (HP-β-CD) | |
|---|---|---|---|---|---|---|
| TCE@β-CD | −4.79 | −26.33 | −25.20 | 0.08 | 0.49 | - |
| TCE@HP-β-CD | −8.52 | −29.20 | −28.93 | 0.13 | - | 0.89 |
| ΔEb | −3.73 | −2.87 | −3.73 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benmerabet, A.; Bouhadiba, A.; Belhocine, Y.; Rahali, S.; Sbei, N.; Seydou, M.; Boucheriha, I.; Omeiri, I.; Assaba, I.M. DFT Investigation on the Complexation of β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin as Recognition Hosts with Trichloroethylene. Atoms 2023, 11, 153. https://doi.org/10.3390/atoms11120153
Benmerabet A, Bouhadiba A, Belhocine Y, Rahali S, Sbei N, Seydou M, Boucheriha I, Omeiri I, Assaba IM. DFT Investigation on the Complexation of β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin as Recognition Hosts with Trichloroethylene. Atoms. 2023; 11(12):153. https://doi.org/10.3390/atoms11120153
Chicago/Turabian StyleBenmerabet, Ahlem, Abdelaziz Bouhadiba, Youghourta Belhocine, Seyfeddine Rahali, Najoua Sbei, Mahamadou Seydou, Ihsene Boucheriha, Imane Omeiri, and Ibtissem Meriem Assaba. 2023. "DFT Investigation on the Complexation of β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin as Recognition Hosts with Trichloroethylene" Atoms 11, no. 12: 153. https://doi.org/10.3390/atoms11120153
APA StyleBenmerabet, A., Bouhadiba, A., Belhocine, Y., Rahali, S., Sbei, N., Seydou, M., Boucheriha, I., Omeiri, I., & Assaba, I. M. (2023). DFT Investigation on the Complexation of β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin as Recognition Hosts with Trichloroethylene. Atoms, 11(12), 153. https://doi.org/10.3390/atoms11120153