
Relativistic Configuration-Interaction and Perturbation Theory Calculations for Heavy Atoms


Abstract
:1. Introduction
2. CI-MBPT Framework
2.1. CI-MBPT Formalism
2.2. CI-MBPT Numerical Procedure
2.3. Ab Initio CI-MBPT
2.4. Ab Initio Relativistic CI
2.5. Parametric CI-MBPT
2.5.1. Energies of Even Th I States
2.5.2. Energies and Transitions of La II and La I
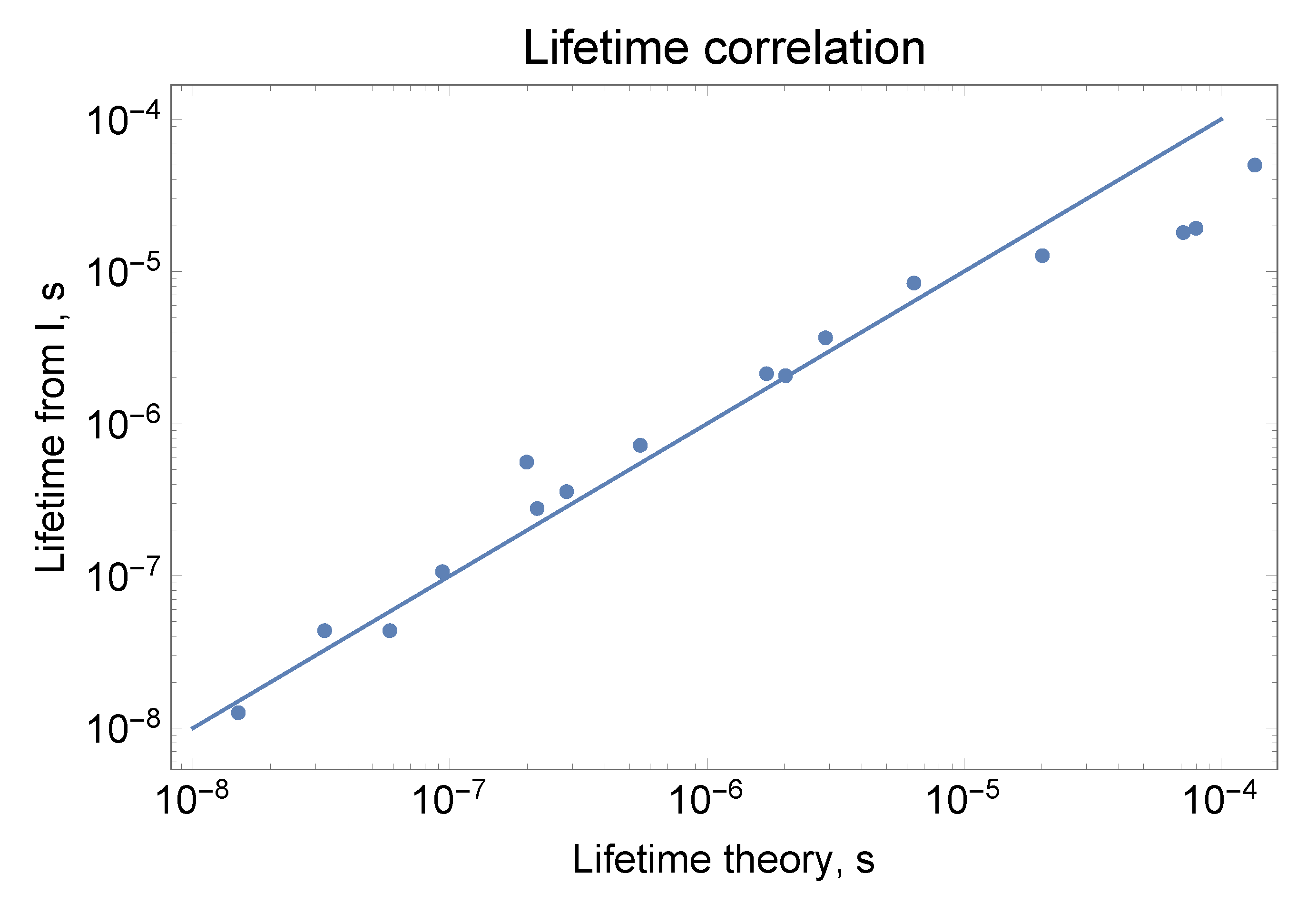
2.5.3. Lifetimes of Th II
2.5.4. U I
2.6. Hyperfine Constant Calculations
2.7. Isotope Shift
2.8. Isotope Shift of Pu II
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savukov, I.M. Parametric CI+MBPT calculations of Th I energies and g-factors for even states. J. Phys. B At. Mol. Opt. Phys. 2017, 50, 165001. [Google Scholar] [CrossRef] [Green Version]
- Savukov, I.M.; Anisimov, P.M. Configuration-interaction many-body perturbation theory for La ii electric-dipole transition probabilities. Phys. Rev. A 2019, 99, 032507. [Google Scholar] [CrossRef] [Green Version]
- Filin, D.; Savukov, I.M. Accurate CI-MBPT calculation of radiative lifetimesand transition probabilities of neutral lanthanum(La I) odd states with J = 3/2. Phys. Scr. 2020, 95, 105401. [Google Scholar] [CrossRef]
- Safronova, M.S.; Safronova, U.I.; Clark, C.W. Relativistic all-order calculations of Th, Th+, and Th2+ atomic properties. Phys. Rev. A 2014, 90, 032512. [Google Scholar] [CrossRef] [Green Version]
- Savukov, I.; Safronova, U.I.; Safronova, M.S. Relativistic configuration interaction plus linearized-coupled-cluster calculations of U2+ energies, g factors, transition rates, and lifetimes. Phys. Rev. A 2015, 92, 052516. [Google Scholar] [CrossRef] [Green Version]
- Savukov, I.M. CI+MBPT calculations of Ar I energies, g factors, and transition line strengths. J. Phys. B At. Mol. Opt. Phys. 2018, 51, 065006. [Google Scholar] [CrossRef]
- Fontes, C.J.; Zhang, H.L.; Abdallah, J., Jr.; Clark, R.E.H.; Kilcrease, D.P.; Colgan, J.; Cunningham, R.T.; Hakel, P.; Magee, N.H.; Sherrill, M.E. The Los Alamos suite of relativistic atomic physics codes. J. Phys. B At. Mol. Opt. Phys. 2015, 48, 144014. [Google Scholar] [CrossRef]
- Liu, H.; Quentmeier, A.; Niemax, K. Diode laser absorption measurement of uranium isotope ratios in solid samples using laser ablation. Spectrochim. Acta Part B 2002, 57, 1611. [Google Scholar] [CrossRef]
- Quentmeier, A.; Bolshov, M.; Niemax, K. Measurements of uranium isotope ratios in solid samples using laser ablation and diode laser-atomic absorption spectroscopy. Spectrochim. Acta Part B 2001, 56, 45. [Google Scholar] [CrossRef]
- Lebedev, V.; Bartlett, J.H.; Castro, A. Isotope-resolved atomic beam laser spectroscopy of natural uranium. J. Anal. At. Spectrom. 2018, 33, 1862–1866. [Google Scholar] [CrossRef]
- Campbell, K.R.; Judge, E.J.; James, E.; Barefield, J.E., II; Colgan, J.P.; Kilcrease, D.P.; Czerwinski, K.R.; Clegg, S.M. Laser-induced spectroscopy of light water reactor simulated used nuclear fuel: Main oxide phase. Spectrochim. Acta Part B 2017, 133, 26. [Google Scholar] [CrossRef]
- Cayrel, R.; Hill, V.; Beers, T.C.; Barbuy, B.; Spite, M.; Spite, F.; Plez, B.; Andersen, J.; Bonifacio, P.; François, P.; et al. Measurement of stellar age from uranium decay. Nature 2001, 409, 691. [Google Scholar] [CrossRef] [Green Version]
- Blaise, J.; Wyart, J.-F. Energy Levels and Atomic Spectra of Actinides. Available online: http://www.lac.universite-paris-saclay.fr/Data/Database/Tab-energy/Uranium/U-tables/U1o-1.html (accessed on 29 November 2021).
- Blaise, J.; Fred, M.; Gutmacher, R.G. Atomic Spectrum of Plutonium; Argonne National Laboratory Report ANL-83-95; Argonne National Lab.: Argonne, IL, USA, 1984; p. 612. [CrossRef] [Green Version]
- Blaise, J.; Fred, M.; Gutmacher, R.G. Term analysis of the spectrum of neutral plutonium, Pu i. J. Opt. Soc. Am. B 1986, 3, 403–418. [Google Scholar] [CrossRef]
- Dzuba, V.A.; Flambaum, V.V. Exponential Increase of Energy Level Density in Atoms: Th and Th II. PRL 2010, 104, 213002. [Google Scholar] [CrossRef] [Green Version]
- Porsev, S.G.; Flambaum, V.V. Electronic bridge process in 229Th+. Phys. Rev. A 2010, 81, 042516. [Google Scholar] [CrossRef] [Green Version]
- Corliss, C.H.; Bozman, W.R. Experimental Transition Probabilities for Spectral Line of Seventy Elements. In NBS Monograph; US Govt Printing Office: Washington, DC, USA, 1962; Volume 53. [Google Scholar]
- Dzuba, V.A. Calculation of the energy levels of Ge, Sn, Pb, and their ions in the VN-4 approximation. Phys. Rev. A 2005, 71, 062501. [Google Scholar] [CrossRef] [Green Version]
- Dzuba, V.A.; Flambaum, V.V.; Silvestrov, P.G.; Sushkov, O.P. Calculation of parity non-conservation in thallium. J. Phys. B 1987, 20, 3297. [Google Scholar] [CrossRef]
- Dzuba, V.A.; Flambaum, V.V.; Sushkov, O.P. Summation of the perturbation theory high order contributions to the correlation correction for the energy levels of the caesium atom. Phys. Lett. A 1989, 140, 493. [Google Scholar] [CrossRef]
- Dzuba, V.A.; Flambaum, V.V.; Kozlov, M.G. Combination of the many-body perturbation theory with the configuration-interaction method. Phys. Rev. A 1996, 54, 3948. [Google Scholar] [CrossRef] [Green Version]
- Savukov, I.M. Configuration-interaction many-body-perturbation-theory energy levels of four-valent Si i. Phys. Rev. A 2015, 91, 022514. [Google Scholar] [CrossRef] [Green Version]
- Savukov, I.M. Configuration-interaction plus many-body-perturbation-theory calculations of Si i transition probabilities, oscillator strengths, and lifetimes. Phys. Rev. A 2016, 93, 022511. [Google Scholar] [CrossRef] [Green Version]
- Savukov, I.M. Relativistic configuration-interaction and many-body-perturbation-theory calculations of U I hyperfine constants. Phys. Rev. A 2020, 102, 042806. [Google Scholar] [CrossRef]
- Kennedy, J. Bare Bones Particle Swarms. In Proceedings of the 2003 IEEE Swarm Intelligence Symposium, Indianapolis, IN, USA, 26 April 2003. [Google Scholar]
- Yang, X.S.; Deb, S.; Fong, S. Accelerated Particle Swarm Optimization and Support Vector Machine for Business Optimization and Applications, NDT 2011; Springer CCIS 136; Springer: Berlin/Heidelberg, Germany, 2011; pp. 53–66. Available online: https://link.springer.com/content/pdf/10.1007%2F978-3-642-22185-9_6.pdf (accessed on 23 November 2021).
- Kułaga-Egger, D.; J Migdałek, J. Theoretical radiative lifetimes of levels in singly ionized lanthanum. J. Phys. B At. Mol. Opt. Phys. 2009, 42, 185002. [Google Scholar] [CrossRef]
- Gamrath, S.; Palmeri, P.; Quinet, P. Radiative transition parameters in atomic lanthanum from pseudo-relativistic Hartree-Fock and fully relativistic Dirac-Hartree-Fock calculations. Atoms 2019, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Karaçoban, B.; Özdemir, L. Electric dipole transitions for La I(Z=57). Quant. Spectrosc. Radiat. Transf. 2008, 109, 1968–1985. [Google Scholar] [CrossRef]
- Corliss, C.H. Oscillator strengths for lines of ionized thorium (Th II). Mon. Not. R. Astr. Soc. 1979, 189, 607. [Google Scholar] [CrossRef] [Green Version]
- Andersen, T.; Petrakiev Petkov, A. Th II Meanlife and the Solar Thorium Abundance. Astron. Astrophys. 1975, 45, 237. [Google Scholar]
- Simonsen, H.; Worm, T.; Jessen, P.; Poulsen, O. Lifetime Measurements and Absolute Oscillator Strengths for Single Ionized Thorium (ThII). Phys. Scr. 1988, 38, 370–373. [Google Scholar] [CrossRef]
- Nilsson, H.; Zhang, Z.G.; Lundberg, H.; Johansson, S.; Nordström, B. Experimental oscillator strengths in Th II. Astron. Astrophys. 2002, 382, 368. [Google Scholar] [CrossRef] [Green Version]
- Gamrath, S.; Godefroid, M.R.; Palmeri, P.; Quinet, P.; Wang, K. Spectral line list of potential cosmochronological interest deduced from new calculations of radiative transition rates in singly ionized thorium (Th II). Mon. Not. R. Astron. Soc. 2020, 496, 4507. [Google Scholar] [CrossRef]
- Peik, E.; Tamm, C. Nuclear laser spectroscopy of the 3.5 eV transition in Th-229. Europhys. Lett. 2003, 61, 181. [Google Scholar] [CrossRef] [Green Version]
- Kerber, F.; Nave, G.; Sansonetti, C.J. The Spectrum of Th-Ar Hollow Cathode Lamps in the 691-5804 nm Region: Establishing Wavelength Standards Calibration of Infrared Spectrographs. Astrophys. J. Suppl. Ser. 2008, 178, 374–381. [Google Scholar] [CrossRef]
- Savukov, I.M. CI-MBPT and intensity-based Lifetime Calculations for ThII. Atoms 2020, 8, 87. [Google Scholar] [CrossRef]
- Avril, R.; Ginibre, A.; Petit, A. On the hyperfine structure in the configuration 5f36d7s2 of neutral uranium. Z. Phys. D 1994, 29, 91–102. [Google Scholar] [CrossRef]
- Rajnak, K.; Fred, M. Correlation of isotope shifts with |ψ(0)|2 for actinide configurations. J. Opt. Soc. Am. 1977, 67, 1314–1323. [Google Scholar] [CrossRef]
- Wilson, M. Ab Initio Calculation of Screening Effects on |ψ(0)|2 for Heavy Atoms. Phys. Rev. 1968, 176, 58–63. [Google Scholar] [CrossRef]
- Cowan, R.D. Atomic self-consistent-field calculations using statistical approximations for exchange and correlation. Phys. Rev. 1967, 163, 54–61. [Google Scholar] [CrossRef]
- Savukov, I. Configuration–Interaction Perturbation Theory Calculations of Pu II. Atoms 2020, 8, 39. [Google Scholar] [CrossRef]
- Rollefson, G.K.; Dodgen, H.W. Report on Spectrographic Analysis Work CK-812; Declassified 29 December 1954; ACS Publication: Washington, DC, USA, July 1943. [Google Scholar]
- McNally, J.R., Jr.; Griffin, P.M. Preliminary classification of the singly ionized plutonium atom (Pu ii). J. Opt. Soc. Am. 1959, 49, 162–166. [Google Scholar] [CrossRef]
- Bovey, L.; Gerstenkorn, S.J. Ground state of the first spectrum of plutonium (Pu i), from an analysis of its atomic spectrum. Opt. Soc. Am. 1961, 51, 522–525. [Google Scholar] [CrossRef]
- Bovey, L.; Ridgeley, A. The Zeeman Effect of Pu I. A.E.R.E.-R3393; Harwell: Oxford, UK, 1960. [Google Scholar]
- Striganov, A.R.; Korostyleva, L.A.; Dontsov, I.P. Isotopic shift in the spectrum of plutonium. Zh. Eksp. Teor. Fiz. 1955, 28, 480, Russian Journal Translation Sov. Phys. JETP 1955, 1, 354. [Google Scholar]
- Korostyleva, L.A. Hyperfine and Isotopic Structure in the Spectrum of Plutonium. Opt. Spectrosc. USSR 1963, 14, 93. Available online: https://ui.adsabs.harvard.edu/abs/1963OptSp..14...93K/abstract (accessed on 21 July 2020).
- Dzuba, V.A.; Johnson, W.R.; Safronova, M.S. Calculation of isotope shifts for cesium and francium. Phys. Rev. A 2005, 72, 022503. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Potential | Coeff.: | |||||||
| , L = 0, L = 1 | ||||||||
| A | −0.0478, 0.0000, 0.4770, 0.0000, 0.6547 | |||||||
| 0.0000, 0.0903, 0.9506, 1.3276, 0.6 | ||||||||
| Conf.DB | Conf.th | g | ||||||
| 0 | 0 | 0 | 0.925 | 22 | 4485 | 4112 | ||
| 3528 | 3525 | 3 | 1.020 | 16 | 1363 | 1239 | ||
| 4336 | 4317 | 19 | 0.845 | 5 | 1661 | 2412 | ||
| 6274 | 6268 | 5 | 0.930 | 94 | 791 | 2744 | ||
| 7791 | 7876 | −85 | 1.095 | −132 | 2861 | |||
| 9081 | 9025 | 56 | 0.890 | 206 | 181 | |||
| 9435 | 9546 | −111 | 0.985 | 171 | −297 | |||
| 9919 | 9767 | 151 | 1.010 | 19 | 2115 | |||
| Potential | Coeff.: | |||||||
| , L = 0, L = 1 | ||||||||
| A | −0.0602, 0.0000, 0.1, 0.0000, 0.2756 | |||||||
| 0.0000, 0.0904, 0.9868, 1.4277, 1.7654 | ||||||||
| Conf.DB | Conf.th | g | ||||||
| 0 | 0 | 0 | 0.925 | 3 | 4578 | 4122 | ||
| 3692 | 3525 | 167 | 1.020 | 27 | 1089 | 1239 | ||
| 4359 | 4317 | 42 | 0.845 | −28 | 1817 | 2412 | ||
| 6745 | 6268 | 477 | 0.930 | 22 | 3163 | 2744 | ||
| 7619 | 7876 | −258 | 1.095 | −99 | 1079 | |||
| 8742 | 9025 | −283 | 0.890 | 228 | 762 | |||
| 9542 | 9546 | −4 | 0.985 | 138 | 377 | |||
| 10,029 | 9767 | 262 | 1.010 | 98 | 230 | |||
| Potential | Coeff.: | |||||||
| , L = 0, L = 1 | ||||||||
| B | 1.8799, 0.1266, 0.4544, 0.2025, 0.4492 | |||||||
| 0.0000, 0.1094, 1.0078, 1.4146, 0.6032 | ||||||||
| Conf.DB | Conf.th | g | ||||||
| 0 | 0 | 0 | 0.925 | 0 | 3543 | 4122 | ||
| 3185 | 3525 | −341 | 1.020 | 27 | 1210 | 1239 | ||
| 4516 | 4317 | 198 | 0.845 | −32 | 2024 | 2412 | ||
| 6681 | 6268 | 412 | 0.930 | −14 | 3151 | 2744 | ||
| 7747 | 7876 | −130 | 1.095 | 42 | 211 | |||
| 8238 | 9025 | −788 | 0.890 | 95 | 1351 | |||
| 9661 | 9546 | 115 | 0.985 | 106 | 604 | |||
| 10,378 | 9767 | 610 | 1.010 | 93 | 192 | |||
| Potential | Coeff.: | |||||||
| , L = 0, L = 1 | ||||||||
| C | 0.5522, 0.6029, 0.6583, 0.8514, 0.7550 | |||||||
| 0.9406, 1.0139, 1.0460, 1.2340, 0.9068 | ||||||||
| Conf.DB | Conf.th | g | ||||||
| 0 | 0 | 0 | 0.925 | −10 | 4100 | 4122 | ||
| 2827 | 3525 | −699 | 1.02 | 30 | 1355 | 1239 | ||
| 4246 | 4317 | −72 | 0.845 | −33 | 2855 | 2412 | ||
| 6516 | 6268 | 248 | 0.93 | 198 | 694 | 2744 | ||
| 7410 | 7876 | −466 | 1.095 | −187 | 3487 | |||
| 8734 | 9025 | −291 | 0.89 | 300 | 228 | |||
| 9620 | 9546 | 74 | 0.985 | −18 | 1907 | |||
| 10,263 | 9767 | 496 | 1.01 | 88 | 990 | |||
| S. No | DB | pRCI | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0.75 | 0.7415 | 0 | ||
| 2 | 4276 | 3905 | 371 | 0.92 | 0.9177 | 25 | ||
| 3 | 6249 | 6247 | 2 | 0.625 | 0.6075 | −545 | ||
| 4 | 7006 | 7253 | −247 | 0.95 | 0.9651 | 5 | ||
| 5 | 10,289 | 9389 | 900 | 1.035 | 1.0568 | 37 | ||
| 6 | 10,988 | 10,629 | 359 | 1.035 | 1.0287 | 28 | ||
| 7 | 11,457 | 10,735 | 722 | 0.81 | 0.804 | −550 | ||
| 8 | 12,911 | 12,650 | 261 | 1.015 | 0.9791 | 0 | ||
| 9 | 13,361 | 13,244 | 117 | 1.015 | 1.0322 | −200 | ||
| 10 | 13,403 | 13,388 | 15 | 0.995 | 1.0765 | −285 | ||
| 8 + 9 + 10 | 1.0083 | 1.0293 | ||||||
| 11 | 14,174 | 14,017 | 157 | 1.145 | 1.2097 | 56 | ||
| 12 | 14,544 | 15,216 | −672 | 0.81 | 1.1017 | −503 | ||
| 13 | 15,435 | 15,562 | −127 | 1.05 | 0.969 | −94 | ||
| 14 | 15,804 | 15,900 | −96 | 1.1 | 0.9478 | −404 | ||
| 15 | 15,906 | 16,108 | −202 | 1.1313 | −463 | |||
| 16 | 16,376 | 16,431 | −55 | 1.0097 | −20 | |||
| 17 | ? | 16,847 | 17,040 | −193 | 1.1057 | |||
| 18 | 17,103 | 17,337 | −234 | 1.0627 | −555 | |||
| 19 | 17,573 | 17,570 | 3 | 1.1266 | 35 | |||
| 20 | 18,006 | 17,878 | 128 | 1.0919 | −535 |
| Conf. | |||||||
|---|---|---|---|---|---|---|---|
| 8709.64 | 8710 | 0 | 0.285 | 0.308 | 551 | 555 | |
| 11,504.095 | 10,808 | −696 | 0.846 | 0.859 | 895 | 897 | |
| 14,295.57 | 14,975 | 679 | 0.777 | 0.79 | 547 | 547 | |
| 15,641.105 | 16,525 | 884 | 0.998 | 1.04 | 551 | 562 | |
| 16,499.64 | 17,824 | 1324 | 0.761 | 0.773 | 494 | 510 | |
| 17,532.945 | 18,549 | 1016 | 1.280 | 1.238 | 572 | 571 | |
| 18,927 | 1.328 | 846 | |||||
| 18,720.09 | 19,802 | 1082 | 0.863 | 1.06 | 507 | 490 | |
| 19,277.2 | 20,671 | 1393 | 0.684 | 0.847 | 454 | 457 | |
| 20,689.1 | 21,416 | 727 | 1.167 | 1.27 | 542 | 543 | |
| 22,040 | 0.927 | 485 | |||||
| 22,373 | 0.8857 | 488 | |||||
| 22,834 | 1.2303 | 535 | |||||
| 22,652.035 | 23,366 | 714 | 1.1822 | 1.185 | 535 | 539 | |
| 23,538.65 | 24,767 | 1229 | 1.1069 | 1.47 | 530 | 535 | |
| 23,671.715 | 25,336 | 1665 | 1.6331 | 1.38 | 549 | 514 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savukov, I.M.; Filin, D.; Chu, P.; Malone, M.W. Relativistic Configuration-Interaction and Perturbation Theory Calculations for Heavy Atoms. Atoms 2021, 9, 104. https://doi.org/10.3390/atoms9040104
Savukov IM, Filin D, Chu P, Malone MW. Relativistic Configuration-Interaction and Perturbation Theory Calculations for Heavy Atoms. Atoms. 2021; 9(4):104. https://doi.org/10.3390/atoms9040104
Chicago/Turabian StyleSavukov, Igor M., Dmytro Filin, Pinghan Chu, and Michael W. Malone. 2021. "Relativistic Configuration-Interaction and Perturbation Theory Calculations for Heavy Atoms" Atoms 9, no. 4: 104. https://doi.org/10.3390/atoms9040104
APA StyleSavukov, I. M., Filin, D., Chu, P., & Malone, M. W. (2021). Relativistic Configuration-Interaction and Perturbation Theory Calculations for Heavy Atoms. Atoms, 9(4), 104. https://doi.org/10.3390/atoms9040104