Apolipoprotein E Interferes with IAPP Aggregation and Protects Pericytes from IAPP-Induced Toxicity

 , , ,
, , ,
Abstract
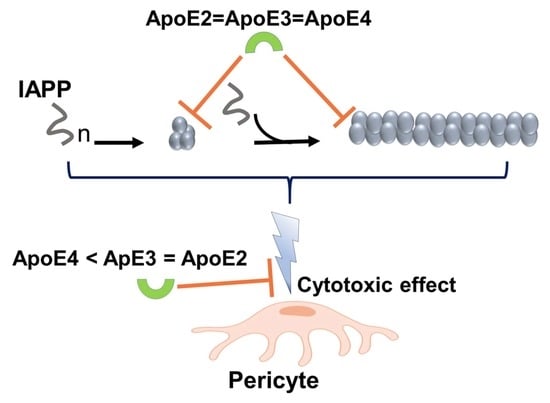
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of ApoE Isoforms and IAPP
2.2. Thioflavin-T Assay
2.3. Negative Staining and Transmission Emission Microscopy
2.4. Cell Viability Assay
3. Results
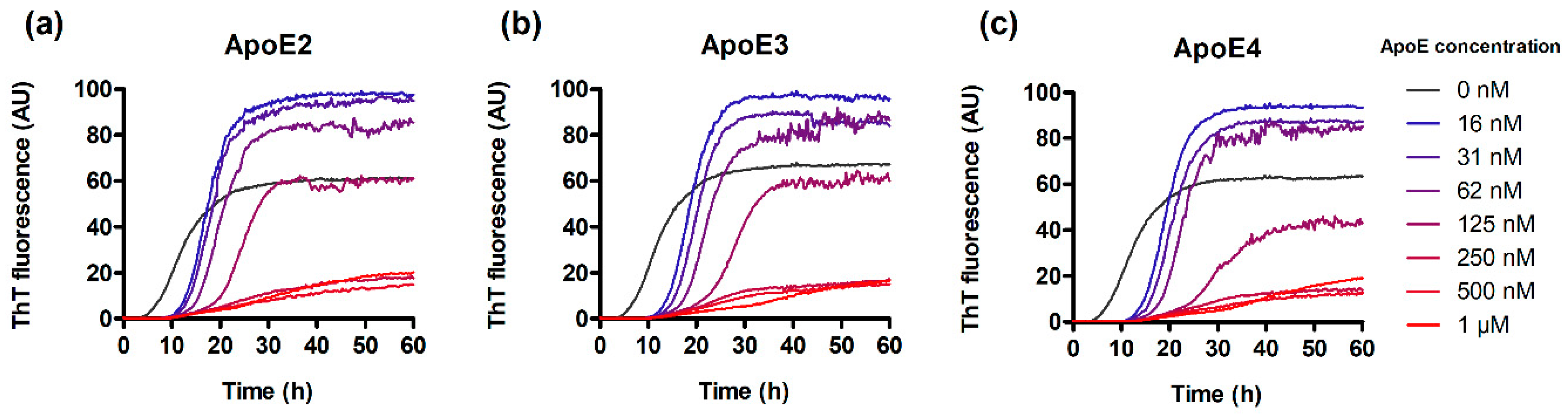
3.1. ApoE Interferes with IAPP Aggregation Kinetics and Suppresses the Formation of Amyloid Structures
3.2. ApoE Changes the Morphology of IAPP Fibrils
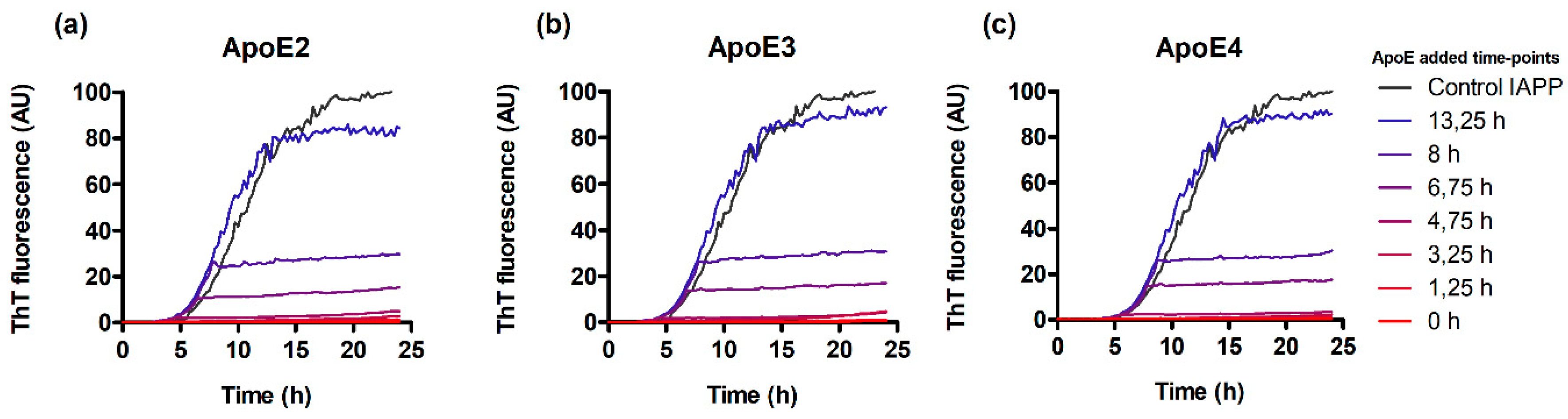
3.3. ApoE Interferes with Nucleation and Elongation during IAPP Amyloid Formation
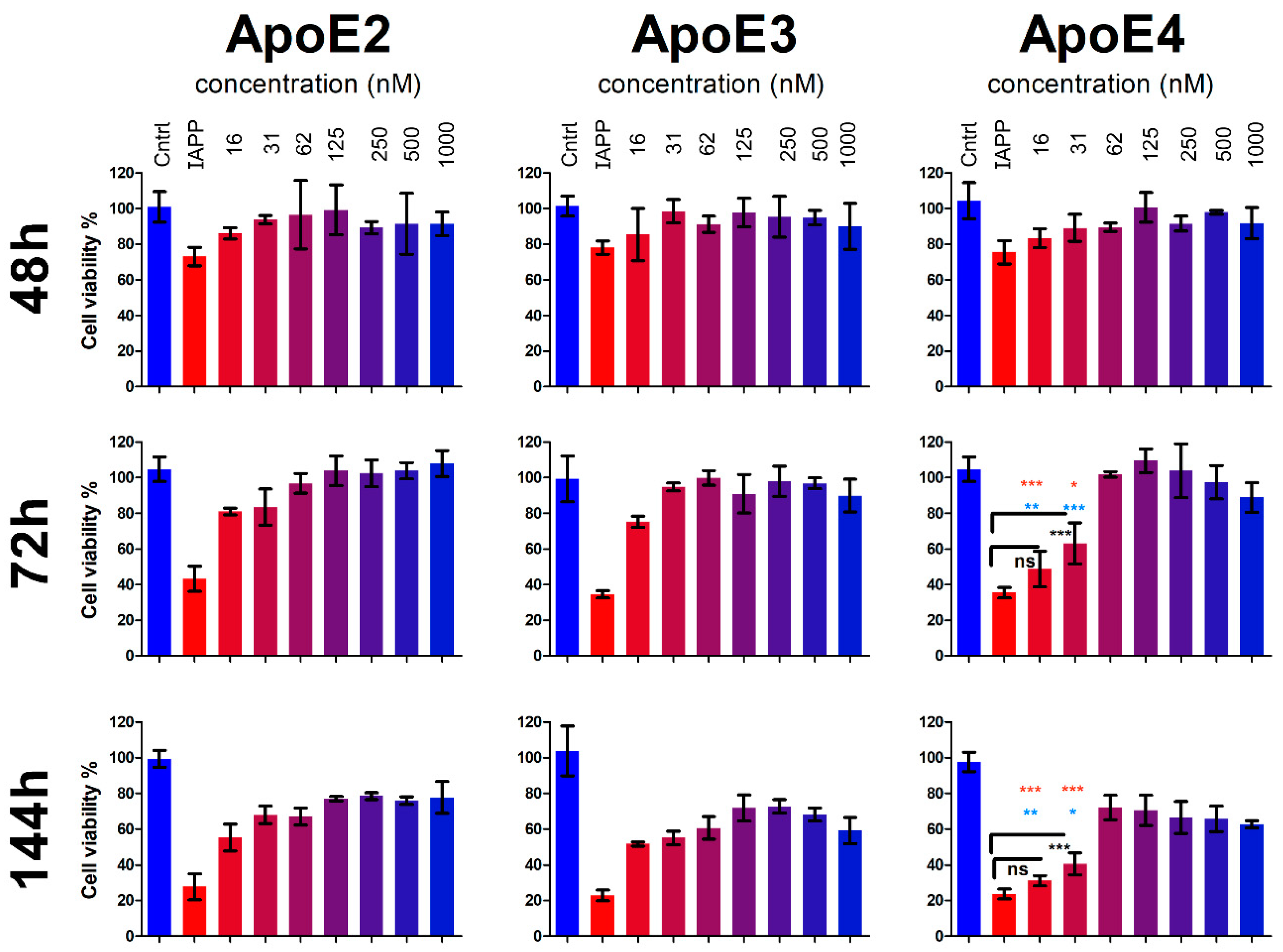
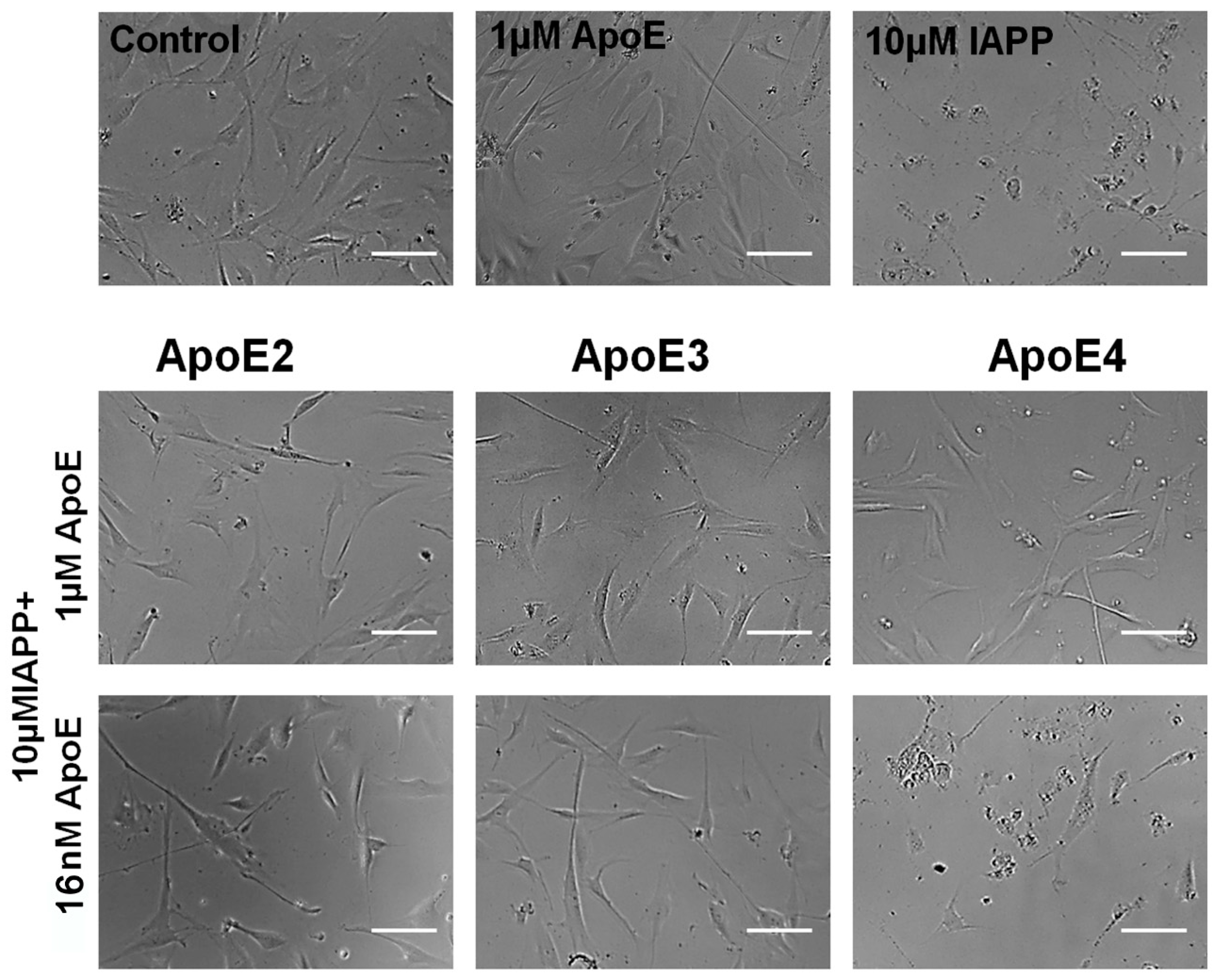
3.4. ApoE Protects the Cells from IAPP-Induced Cytotoxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yu, J.T.; Tan, L.; Hardy, J. Apolipoprotein E in Alzheimer’s disease: An update. Annu. Rev. Neurosci. 2014, 37, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Frangione, B. Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 1992, 135, 235–238. [Google Scholar] [CrossRef]
- Rall, S.C., Jr.; Weisgraber, K.H.; Mahley, R.W. Human apolipoprotein E. The complete amino acid sequence. J. Biol. Chem. 1982, 257, 4171–4178. [Google Scholar]
- Weisgraber, K.H.; Rall, S.C., Jr.; Mahley, R.W. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981, 256, 9077–9083. [Google Scholar]
- Raulin, A.C.; Kraft, L.; Al-Hilaly, Y.K.; Xue, W.F.; McGeehan, J.E.; Atack, J.R.; Serpell, L. The Molecular Basis for Apolipoprotein E4 as the Major Risk Factor for Late-Onset Alzheimer’s Disease. J. Mol. Biol. 2019, 431, 2248–2265. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Hatters, D.M.; Budamagunta, M.S.; Voss, J.C.; Weisgraber, K.H. Modulation of apolipoprotein E structure by domain interaction: Differences in lipid-bound and lipid-free forms. J. Biol. Chem. 2005, 280, 34288–34295. [Google Scholar] [CrossRef] [Green Version]
- Clement-Collin, V.; Barbier, A.; Dergunov, A.D.; Visvikis, A.; Siest, G.; Desmadril, M.; Takahashi, M.; Aggerbeck, L.P. The structure of human apolipoprotein E2, E3 and E4 in solution. 2. Multidomain organization correlates with the stability of apoE structure. Biophys. Chem. 2006, 119, 170–185. [Google Scholar] [CrossRef]
- Hatters, D.M.; Zhong, N.; Rutenber, E.; Weisgraber, K.H. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J. Mol. Biol. 2006, 361, 932–944. [Google Scholar] [CrossRef] [Green Version]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, G.J.; Silvestri, G.; Chakravarthy, U.; Dasari, S.; Fritsche, L.G.; Weber, B.H.; Keilhauer, C.N.; Klein, M.L.; Francis, P.J.; Klaver, C.C.; et al. Variations in apolipoprotein E frequency with age in a pooled analysis of a large group of older people. Am. J. Epidemiol. 2011, 173, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.B.; Adams, K.W.; Rozkalne, A.; Spires-Jones, T.L.; Hshieh, T.T.; Hashimoto, T.; von Armin, C.A.; Mielke, M.; Bacskai, B.J.; Hyman, B.T. Apolipoprotein E: Isoform specific differences in tertiary structure and interaction with amyloid-beta in human Alzheimer brain. PLoS ONE 2011, 6, e14586. [Google Scholar] [CrossRef] [PubMed]
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garai, K.; Ghosh, S.; Sil, T.B.; Dolai, S. Apolipoprotein E interacts with amyloid-β oligomers via positively cooperative multivalent binding. bioRxiv 2018, 473892. [Google Scholar] [CrossRef]
- Lee, C.Y.; Tse, W.; Smith, J.D.; Landreth, G.E. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 2012, 287, 2032–2044. [Google Scholar] [CrossRef] [Green Version]
- Bruinsma, I.B.; Wilhelmus, M.M.; Kox, M.; Veerhuis, R.; de Waal, R.M.; Verbeek, M.M. Apolipoprotein E protects cultured pericytes and astrocytes from D-Abeta(1-40)-mediated cell death. Brain Res. 2010, 1315, 169–180. [Google Scholar] [CrossRef]
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J. Neurosci. 2012, 32, 15181–15192. [Google Scholar] [CrossRef] [Green Version]
- Ly, S.; Altman, R.; Petrlova, J.; Lin, Y.; Hilt, S.; Huser, T.; Laurence, T.A.; Voss, J.C. Binding of apolipoprotein E inhibits the oligomer growth of amyloid-beta peptide in solution as determined by fluorescence cross-correlation spectroscopy. J. Biol. Chem. 2013, 288, 11628–11635. [Google Scholar] [CrossRef] [Green Version]
- Garai, K.; Verghese, P.B.; Baban, B.; Holtzman, D.M.; Frieden, C. The binding of apolipoprotein E to oligomers and fibrils of amyloid-beta alters the kinetics of amyloid aggregation. Biochemistry 2014, 53, 6323–6331. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Yoon, H.; Kim, J. Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr. Opin. Lipidol. 2017, 28, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018, 13, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, H.; Ma, J.; Das, S.; Kayyali, U. The involvement of amyloid associated proteins in the formation of beta-protein filaments in Alzheimer’s disease. Prog. Clin. Biol. Res. 1994, 390, 57–71. [Google Scholar]
- Holtzman, D.M.; Fagan, A.M.; Mackey, B.; Tenkova, T.; Sartorius, L.; Paul, S.M.; Bales, K.; Ashe, K.H.; Irizarry, M.C.; Hyman, B.T. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann. Neurol. 2000, 47, 739–747. [Google Scholar] [CrossRef]
- Mangrolia, P.; Yang, D.T.; Murphy, R.M. Transthyretin variants with improved inhibition of beta-amyloid aggregation. Protein Eng. Des. Sel. 2016, 29, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, L.; Pamren, A.; Islam, T.; Brannstrom, K.; Golchin, S.A.; Pettersson, N.; Iakovleva, I.; Sandblad, L.; Gharibyan, A.L.; Olofsson, A. Transthyretin Interferes with Abeta Amyloid Formation by Redirecting Oligomeric Nuclei into Non-Amyloid Aggregates. J. Mol. Biol. 2018, 430, 2722–2733. [Google Scholar] [CrossRef]
- Willander, H.; Presto, J.; Askarieh, G.; Biverstal, H.; Frohm, B.; Knight, S.D.; Johansson, J.; Linse, S. BRICHOS domains efficiently delay fibrillation of amyloid beta-peptide. J. Biol. Chem. 2012, 287, 31608–31617. [Google Scholar] [CrossRef] [Green Version]
- Beeg, M.; Stravalaci, M.; Romeo, M.; Carra, A.D.; Cagnotto, A.; Rossi, A.; Diomede, L.; Salmona, M.; Gobbi, M. Clusterin Binds to Abeta1-42 Oligomers with High Affinity and Interferes with Peptide Aggregation by Inhibiting Primary and Secondary Nucleation. J. Biol. Chem. 2016, 291, 6958–6966. [Google Scholar] [CrossRef] [Green Version]
- Islam, T.; Gharibyan, A.L.; Golchin, S.A.; Pettersson, N.; Brannstrom, K.; Hedberg, I.; Virta, M.M.; Olofsson, L.; Olofsson, A. Apolipoprotein E impairs amyloid-beta fibril elongation and maturation. FEBS J. 2019. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. Elife 2017, 6, e29595. [Google Scholar] [CrossRef]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef]
- Rezeli, M.; Zetterberg, H.; Blennow, K.; Brinkmalm, A.; Laurell, T.; Hansson, O.; Marko-Varga, G. Quantification of total apolipoprotein E and its specific isoforms in cerebrospinal fluid and blood in Alzheimer’s disease and other neurodegenerative diseases. EuPA Open Proteom. 2015, 8, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.B.; Laws, S.M.; Villemagne, V.L.; Ames, D.; Bush, A.I.; Ellis, K.A.; Lui, J.K.; Masters, C.; Rowe, C.C.; Szoeke, C.; et al. Plasma apolipoprotein E and Alzheimer disease risk: The AIBL study of aging. Neurology 2011, 76, 1091–1098. [Google Scholar] [CrossRef]
- Rasmussen, K.L. Plasma levels of apolipoprotein E, APOE genotype and risk of dementia and ischemic heart disease: A review. Atherosclerosis 2016, 255, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma apolipoprotein E levels and risk of dementia: A Mendelian randomization study of 106,562 individuals. Alzheimers Dement. 2018, 14, 71–80. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [Green Version]
- Gurlo, T.; Ryazantsev, S.; Huang, C.J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for proteotoxicity in beta cells in type 2 diabetes: Toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol. 2010, 176, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Aston-Mourney, K.; Hull, R.L.; Zraika, S.; Udayasankar, J.; Subramanian, S.L.; Kahn, S.E. Exendin-4 increases islet amyloid deposition but offsets the resultant beta cell toxicity in human islet amyloid polypeptide transgenic mouse islets. Diabetologia 2011, 54, 1756–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zraika, S.; Hull, R.L.; Verchere, C.B.; Clark, A.; Potter, K.J.; Fraser, P.E.; Raleigh, D.P.; Kahn, S.E. Toxic oligomers and islet beta cell death: Guilty by association or convicted by circumstantial evidence? Diabetologia 2010, 53, 1046–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Despa, S.; Margulies, K.B.; Chen, L.; Knowlton, A.A.; Havel, P.J.; Taegtmeyer, H.; Bers, D.M.; Despa, F. Hyperamylinemia contributes to cardiac dysfunction in obesity and diabetes: A study in humans and rats. Circ. Res. 2012, 110, 598–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, W.; Liu, Z.H.; Zeng, C.H.; Peng, A.; Chen, H.P.; Zhou, H.; Li, L.S. Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int. 2007, 72, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Ly, H.; Despa, F. Hyperamylinemia as a risk factor for accelerated cognitive decline in diabetes. Expert Rev. Proteom. 2015, 12, 575–577. [Google Scholar] [CrossRef] [Green Version]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Vidal, J.; Verchere, C.B.; Andrikopoulos, S.; Wang, F.; Hull, R.L.; Cnop, M.; Olin, K.L.; LeBoeuf, R.C.; O’Brien, K.D.; Chait, A.; et al. The effect of apolipoprotein E deficiency on islet amyloid deposition in human islet amyloid polypeptide transgenic mice. Diabetologia 2003, 46, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.S.; Maksoud, H.; Chargé, S.B.P.; Moffitt, J.H.; Desai, M.; Da Silva Fihlo, R.L.; Hattersley, A.T.; Stratton, I.M.; Matthews, D.R.; Levy, J.C.; et al. Apolipoprotein E genotype, islet amyloid deposition and severity of Type 2 diabetes. Diabetes Res. Clin. Pract. 2003, 60, 105–110. [Google Scholar] [CrossRef]
- Guan, J.; Zhao, H.L.; Sui, Y.; He, L.; Lee, H.M.; Lai, F.M.; Tong, P.C.; Chan, J.C. Histopathological correlations of islet amyloidosis with apolipoprotein E polymorphisms in type 2 diabetic Chinese patients. Pancreas 2013, 42, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Wu, W.H.; Li, R.W.; Ma, J.W.; Yu, Y.P.; Cui, W.; Zhao, Y.F.; Li, Y.M. Prevention and promotion effects of apolipoprotein E4 on amylin aggregation. Biochem. Biophys. Res. Commun. 2008, 368, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Byman, E.; Fex, M.; Wennstrom, M. Amylin alters human brain pericyte viability and NG2 expression. J. Cereb. Blood Flow Metab. 2017, 37, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Sparr, E.; Engel, M.F.; Sakharov, D.V.; Sprong, M.; Jacobs, J.; de Kruijff, B.; Hoppener, J.W.; Killian, J.A. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004, 577, 117–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009, 52, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, J.F.; Gurlo, T.; Daval, M.; Huang, C.J.; Matveyenko, A.V.; Butler, P.C.; Costes, S. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic beta-cells: Protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. 2011, 18, 415–426. [Google Scholar] [CrossRef]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes. Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Beltramo, E.; Porta, M. Pericyte loss in diabetic retinopathy: Mechanisms and consequences. Curr. Med. Chem. 2013, 20, 3218–3225. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The Pericyte: A Forgotten Cell Type with Important Implications for Alzheimer’s Disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef]
- Godin, E.; Nguyen, P.T.; Zottig, X.; Bourgault, S. Identification of a hinge residue controlling islet amyloid polypeptide self-assembly and cytotoxicity. J. Biol. Chem. 2019, 294, 8452–8463. [Google Scholar] [CrossRef]
- Buchanan, L.E.; Dunkelberger, E.B.; Tran, H.Q.; Cheng, P.N.; Chiu, C.C.; Cao, P.; Raleigh, D.P.; de Pablo, J.J.; Nowick, J.S.; Zanni, M.T. Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient beta-sheet. Proc. Natl. Acad. Sci. USA 2013, 110, 19285–19290. [Google Scholar] [CrossRef] [Green Version]
- Linse, S. Monomer-dependent secondary nucleation in amyloid formation. Biophys. Rev. 2017, 9, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Tornquist, M.; Michaels, T.C.T.; Sanagavarapu, K.; Yang, X.; Meisl, G.; Cohen, S.I.A.; Knowles, T.P.J.; Linse, S. Secondary nucleation in amyloid formation. Chem. Commun. 2018, 54, 8667–8684. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Sil, T.B.; Dolai, S.; Garai, K. High-affinity multivalent interactions between apolipoprotein E and the oligomers of amyloid-beta. FEBS J. 2019, 286, 4737–4753. [Google Scholar] [CrossRef]
- Liu, S.; Park, S.; Allington, G.; Prelli, F.; Sun, Y.; Martá-Ariza, M.; Scholtzova, H.; Biswas, G.; Brown, B.; Verghese, P.B.; et al. Targeting Apolipoprotein E/Amyloid β Binding by Peptoid CPO_Aβ17-21 P Ameliorates Alzheimer’s Disease Related Pathology and Cognitive Decline. Sci. Rep. 2017, 7, 8009. [Google Scholar] [CrossRef] [Green Version]
- Bales, K.R.; Verina, T.; Dodel, R.C.; Du, Y.; Altstiel, L.; Bender, M.; Hyslop, P.; Johnstone, E.M.; Little, S.P.; Cummins, D.J.; et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 1997, 17, 263–264. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gharibyan, A.L.; Islam, T.; Pettersson, N.; Golchin, S.A.; Lundgren, J.; Johansson, G.; Genot, M.; Schultz, N.; Wennström, M.; Olofsson, A. Apolipoprotein E Interferes with IAPP Aggregation and Protects Pericytes from IAPP-Induced Toxicity. Biomolecules 2020, 10, 134. https://doi.org/10.3390/biom10010134
Gharibyan AL, Islam T, Pettersson N, Golchin SA, Lundgren J, Johansson G, Genot M, Schultz N, Wennström M, Olofsson A. Apolipoprotein E Interferes with IAPP Aggregation and Protects Pericytes from IAPP-Induced Toxicity. Biomolecules. 2020; 10(1):134. https://doi.org/10.3390/biom10010134
Chicago/Turabian StyleGharibyan, Anna L., Tohidul Islam, Nina Pettersson, Solmaz A. Golchin, Johanna Lundgren, Gabriella Johansson, Mélany Genot, Nina Schultz, Malin Wennström, and Anders Olofsson. 2020. "Apolipoprotein E Interferes with IAPP Aggregation and Protects Pericytes from IAPP-Induced Toxicity" Biomolecules 10, no. 1: 134. https://doi.org/10.3390/biom10010134
APA StyleGharibyan, A. L., Islam, T., Pettersson, N., Golchin, S. A., Lundgren, J., Johansson, G., Genot, M., Schultz, N., Wennström, M., & Olofsson, A. (2020). Apolipoprotein E Interferes with IAPP Aggregation and Protects Pericytes from IAPP-Induced Toxicity. Biomolecules, 10(1), 134. https://doi.org/10.3390/biom10010134