Lysosomal Fusion: An Efficient Mechanism Increasing Their Sequestration Capacity for Weak Base Drugs without Apparent Lysosomal Biogenesis



Abstract
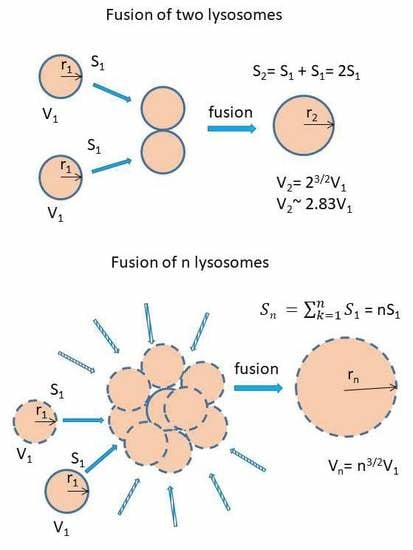
:
{kind=link}
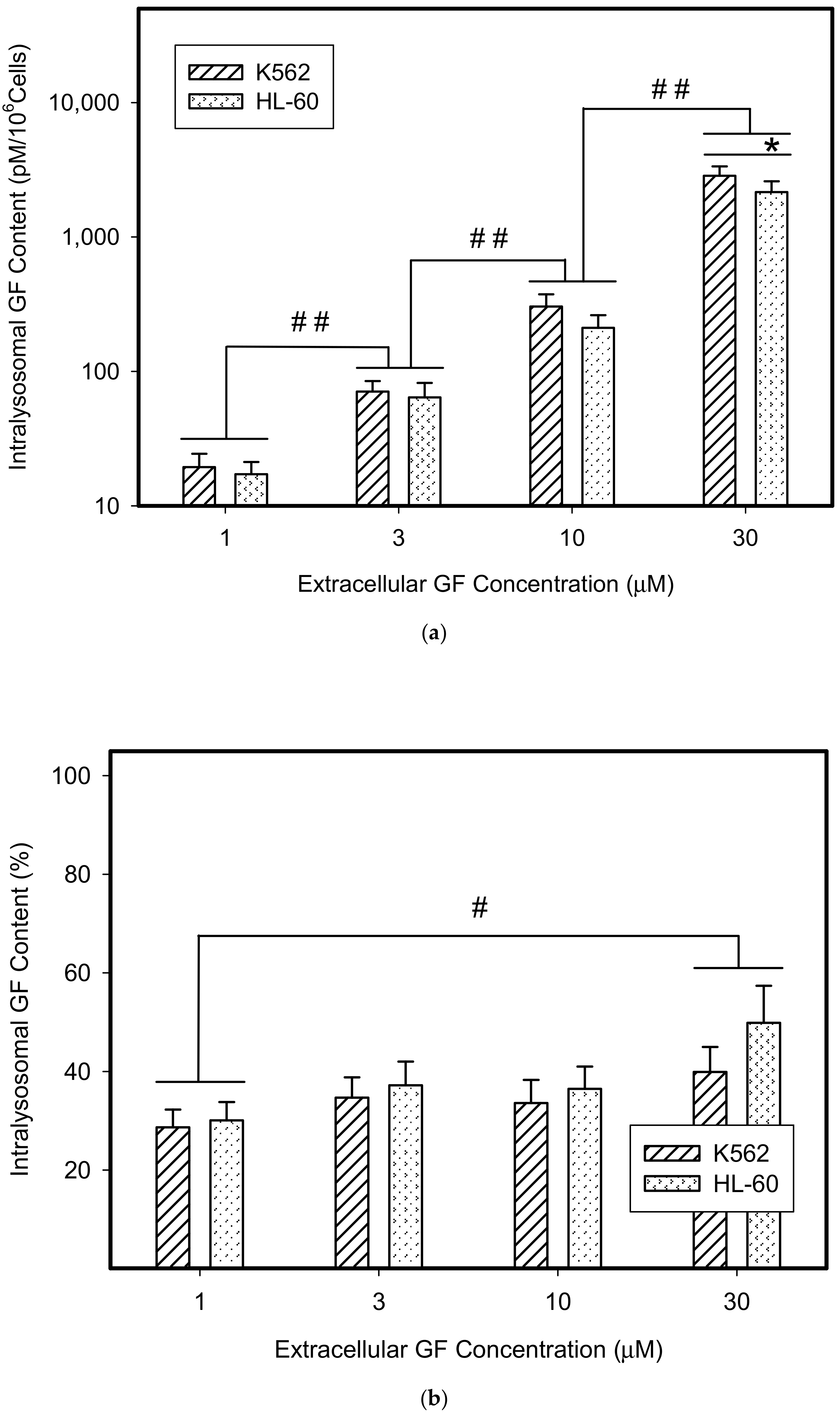{kind=link}
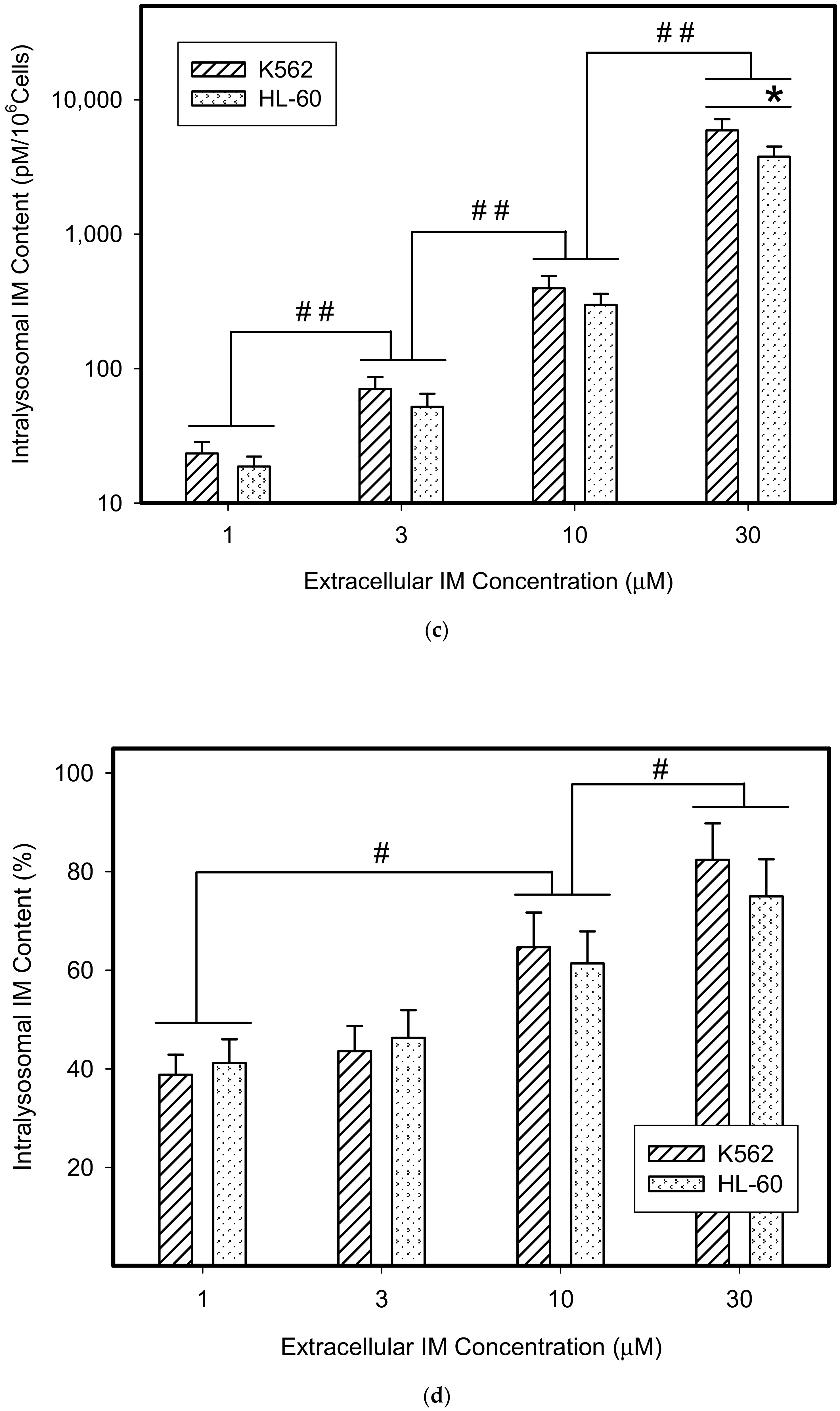{kind=link}
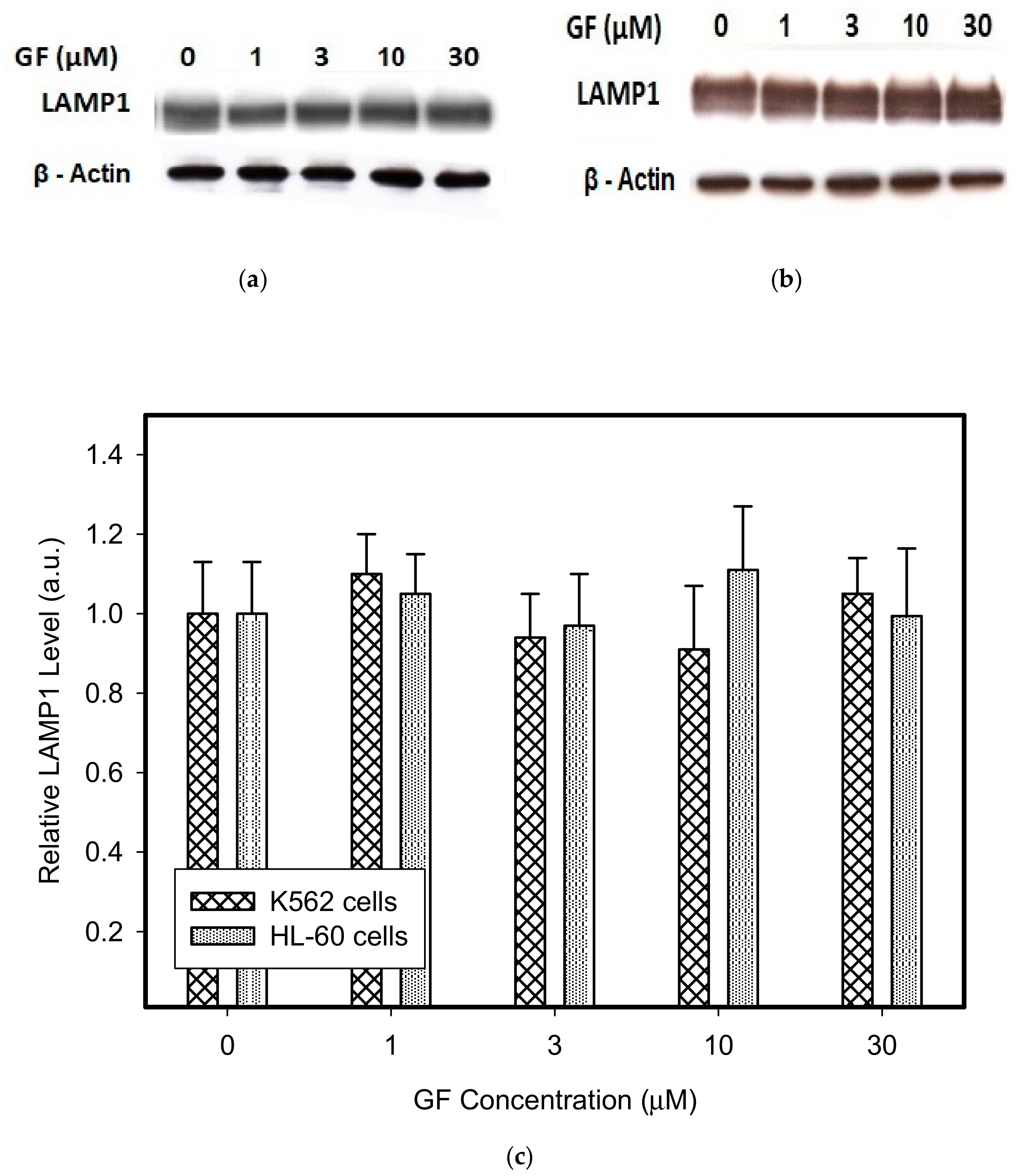{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
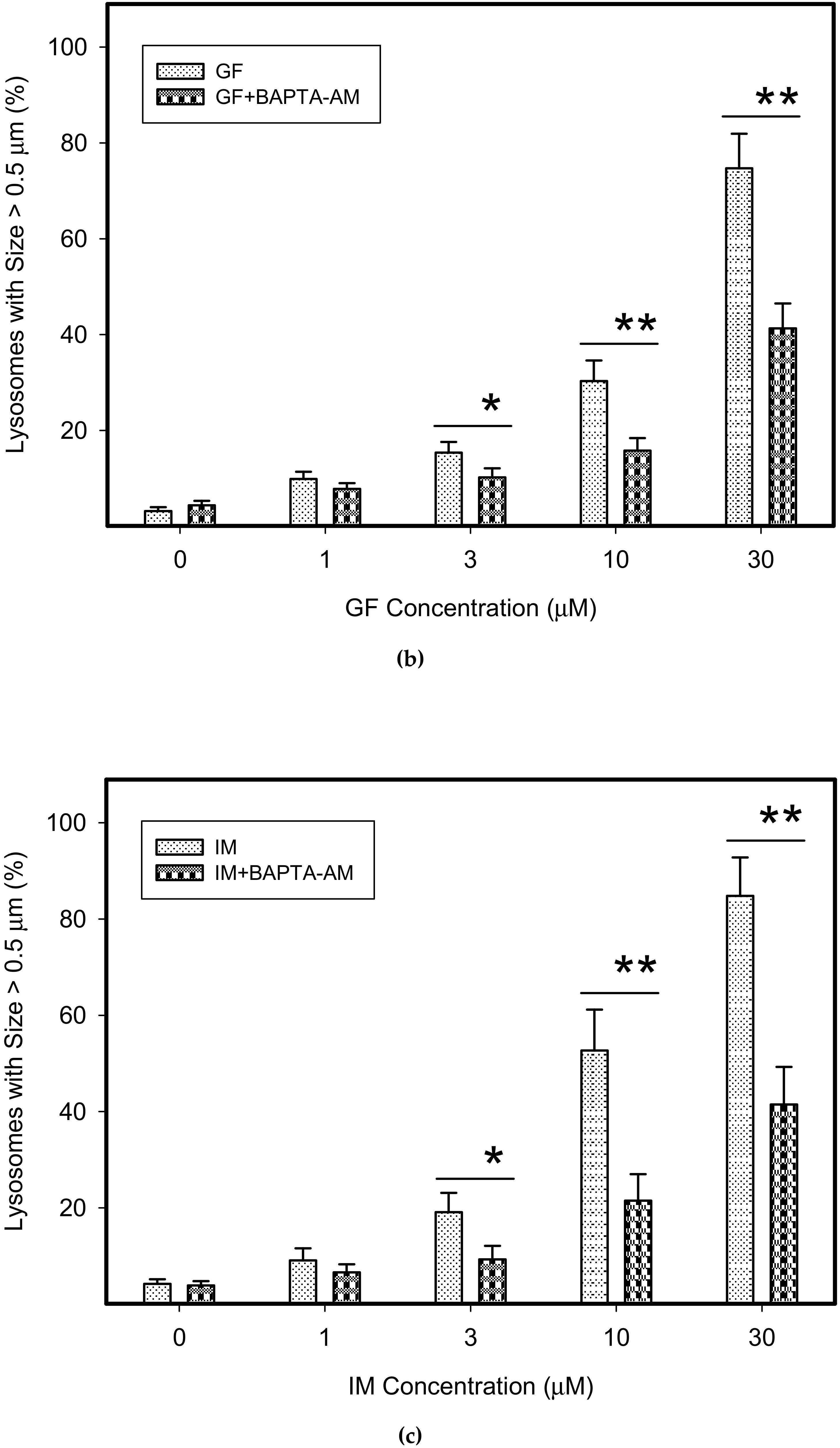{kind=link}
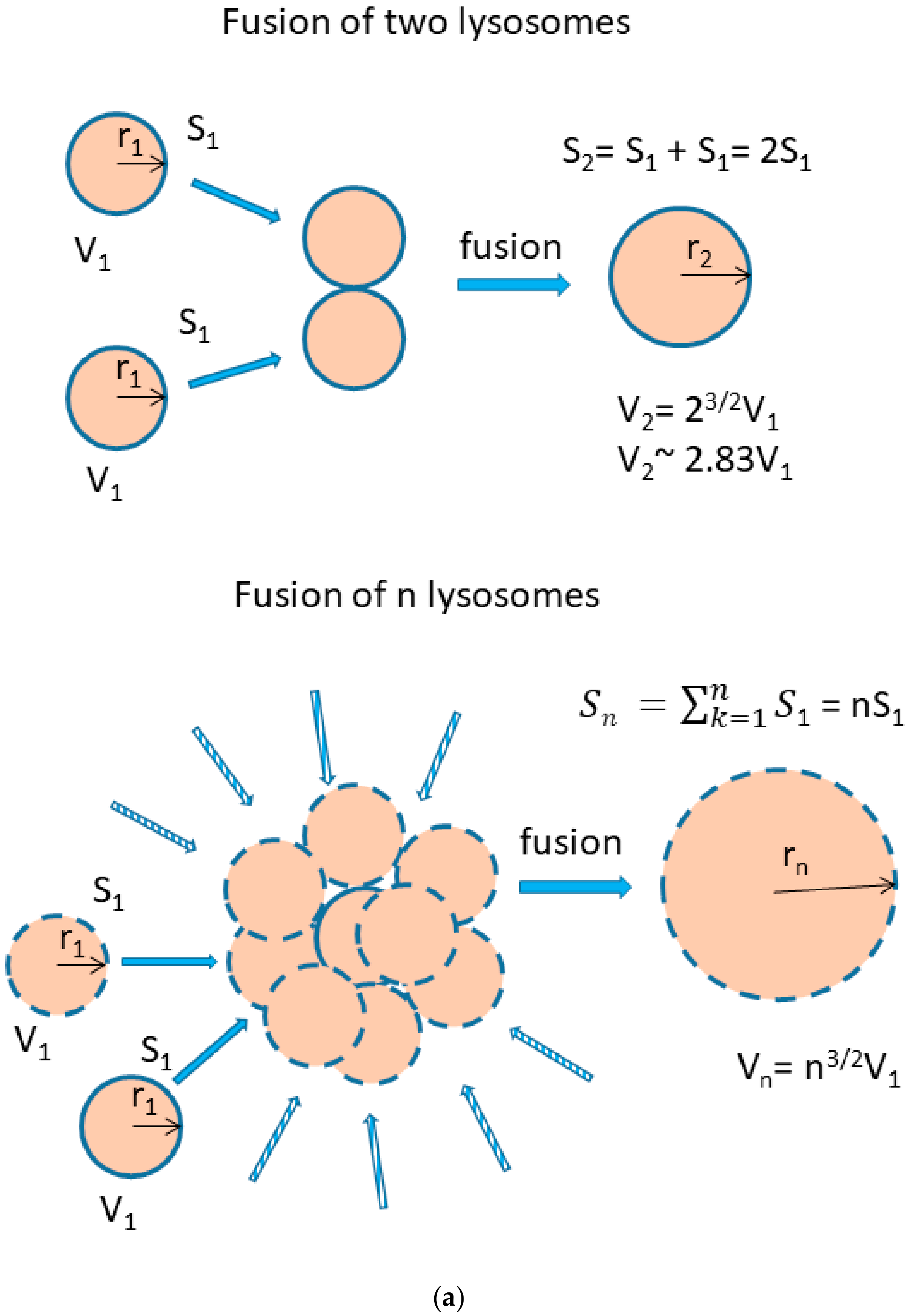{kind=link}
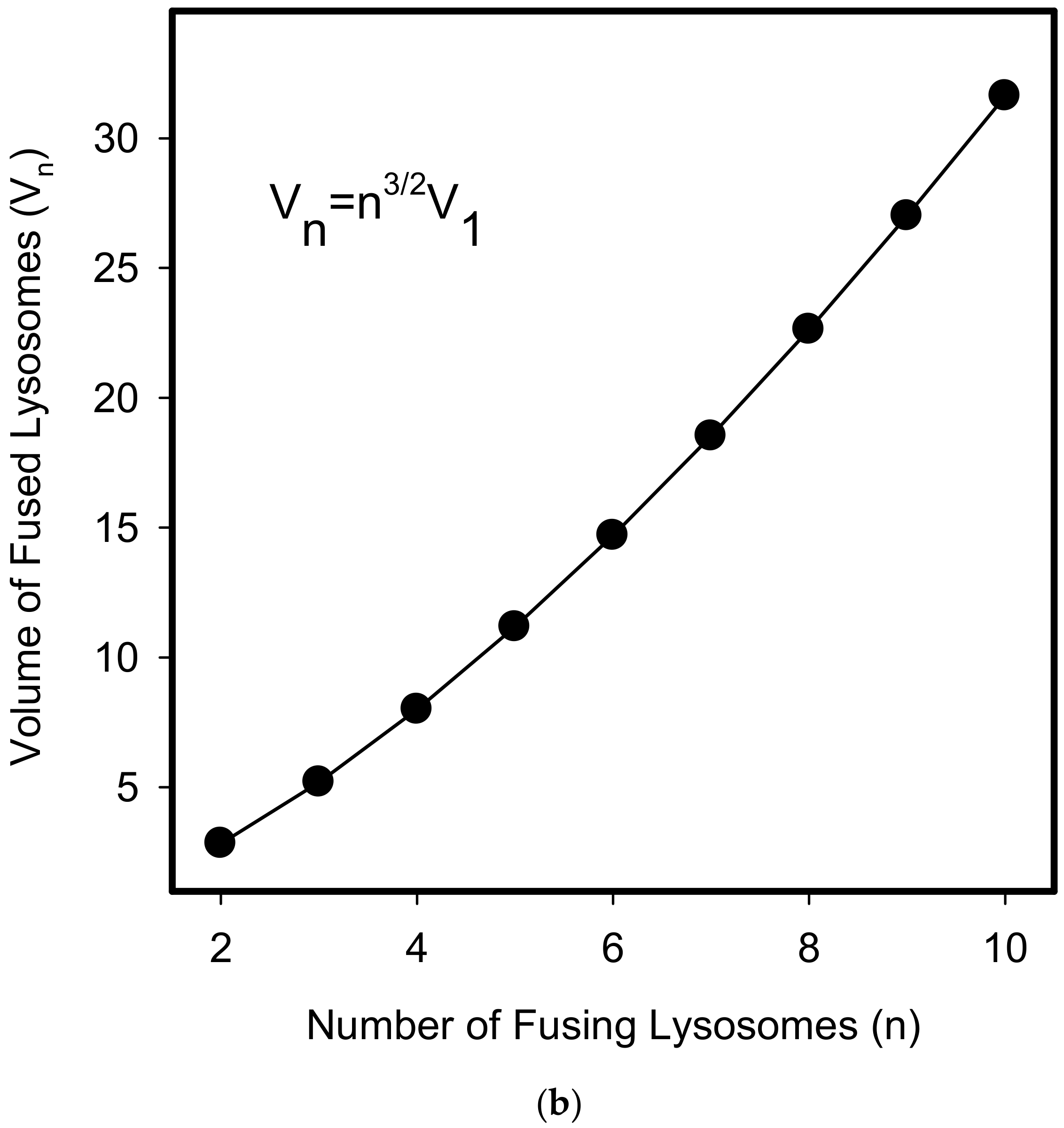{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Assay for Determination of Intracellular IM Levels
2.4. Assay for Determination of Intracellular GF Levels
2.5. Calculation of TKIs in Lysosomes
2.6. Western Blot Analysis
2.7. Activity of Lysosomal Hydrolases
2.8. Immunostaining of LAMP1
2.9. Statistical Analysis
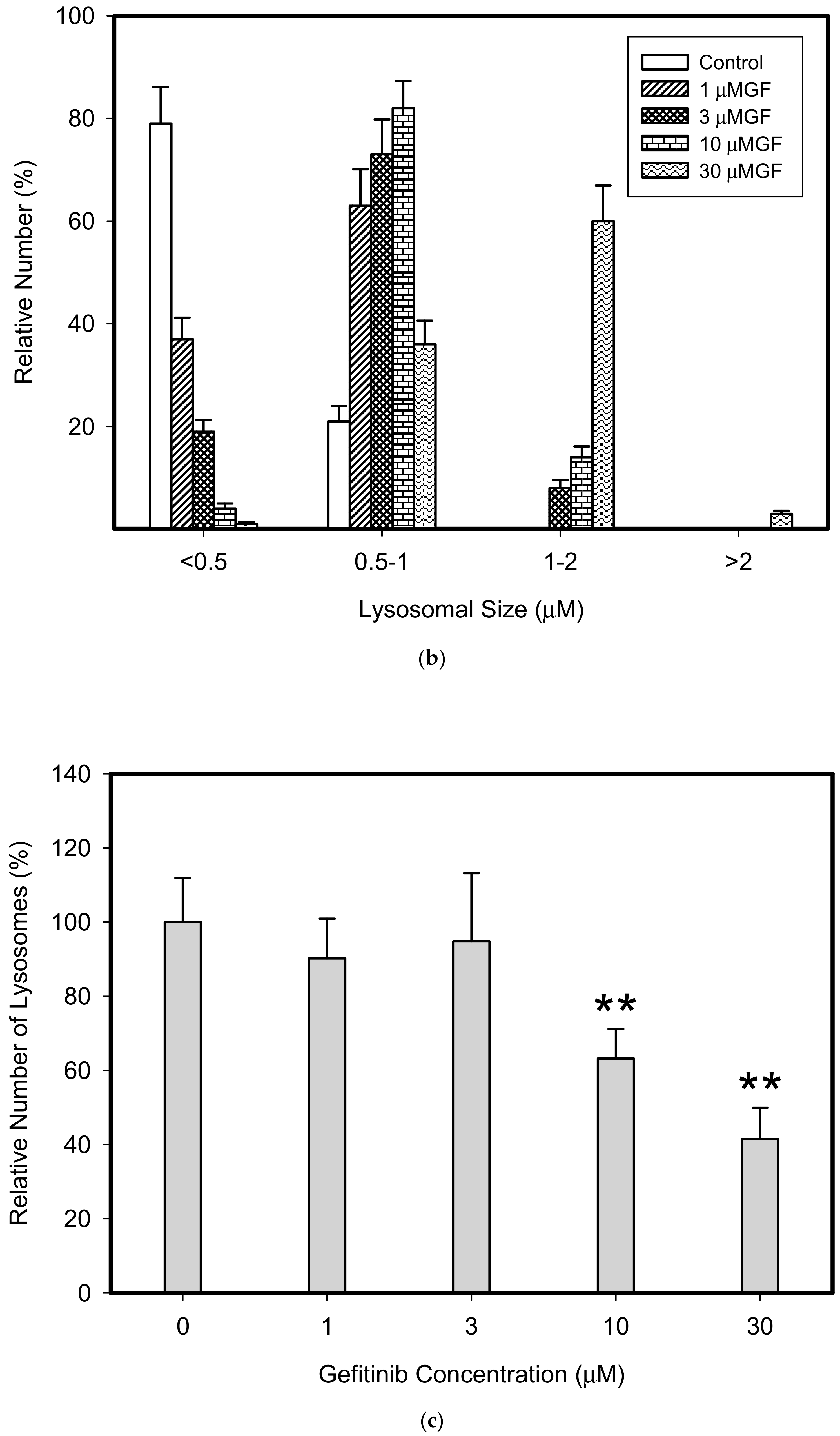
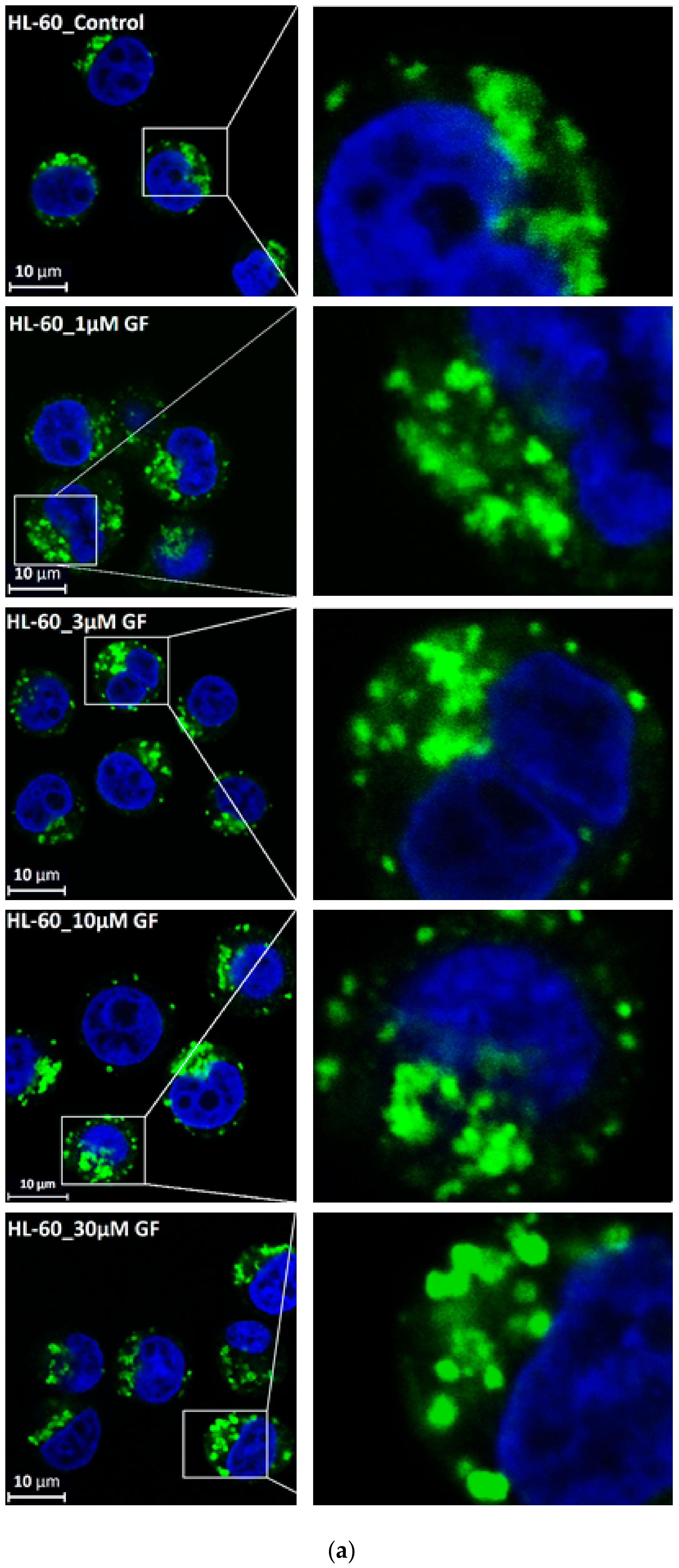
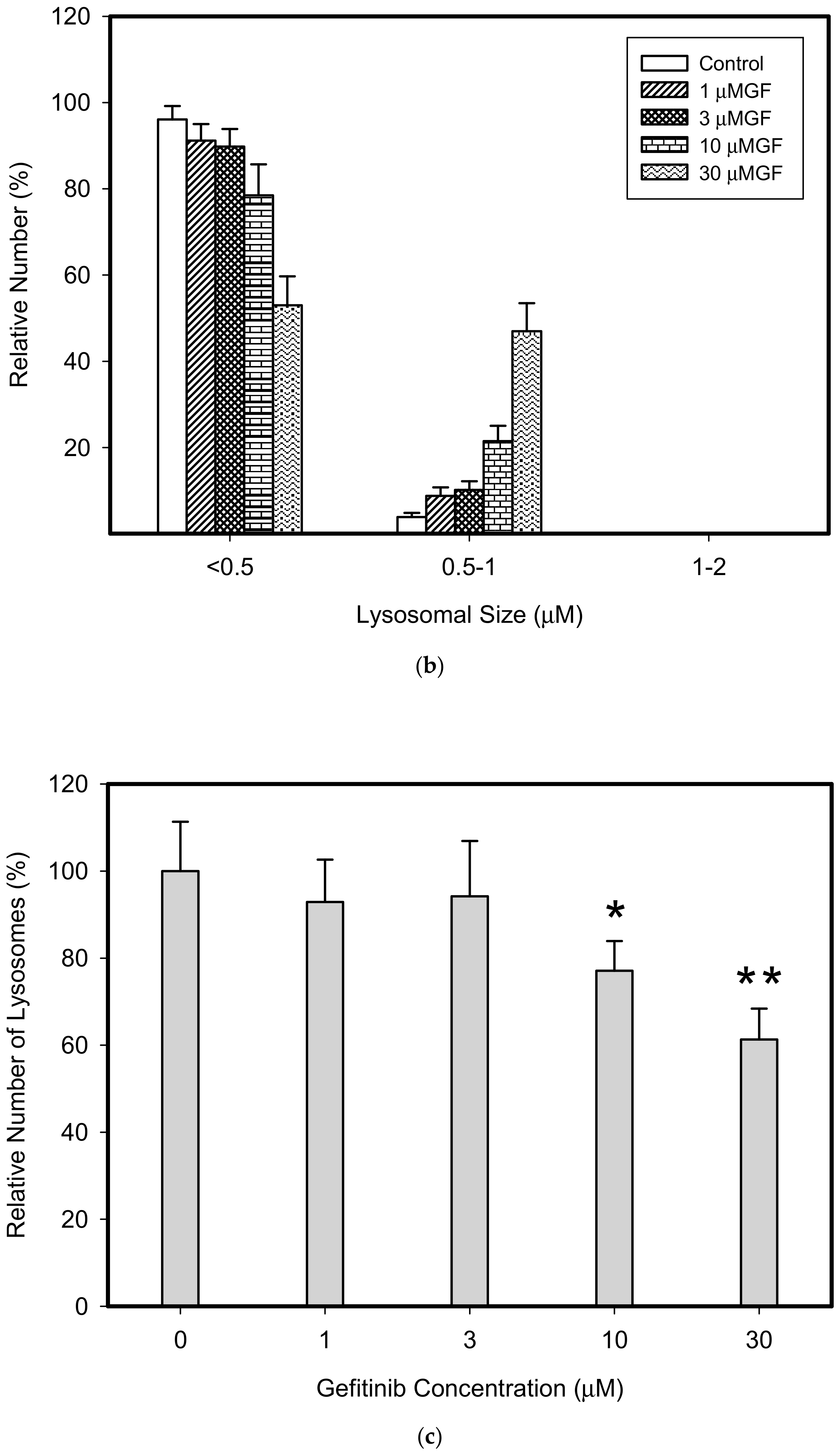
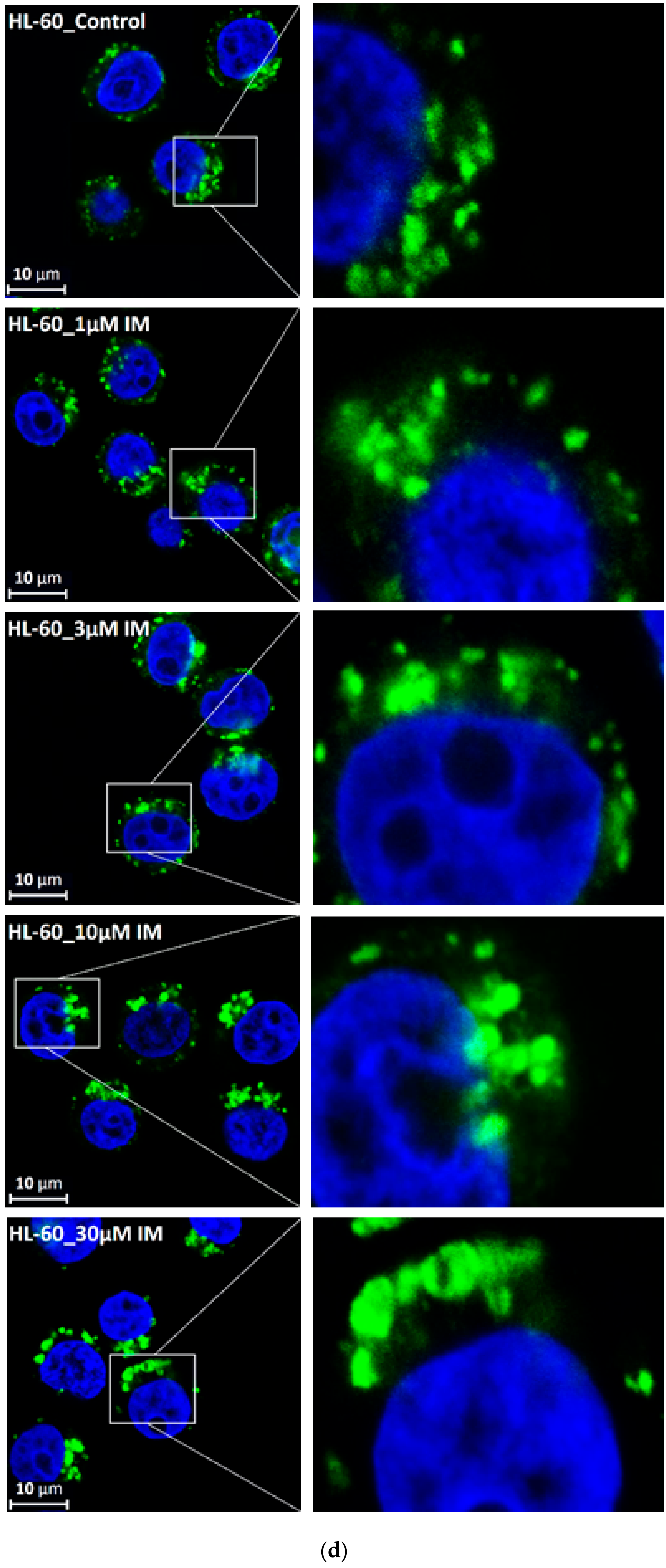
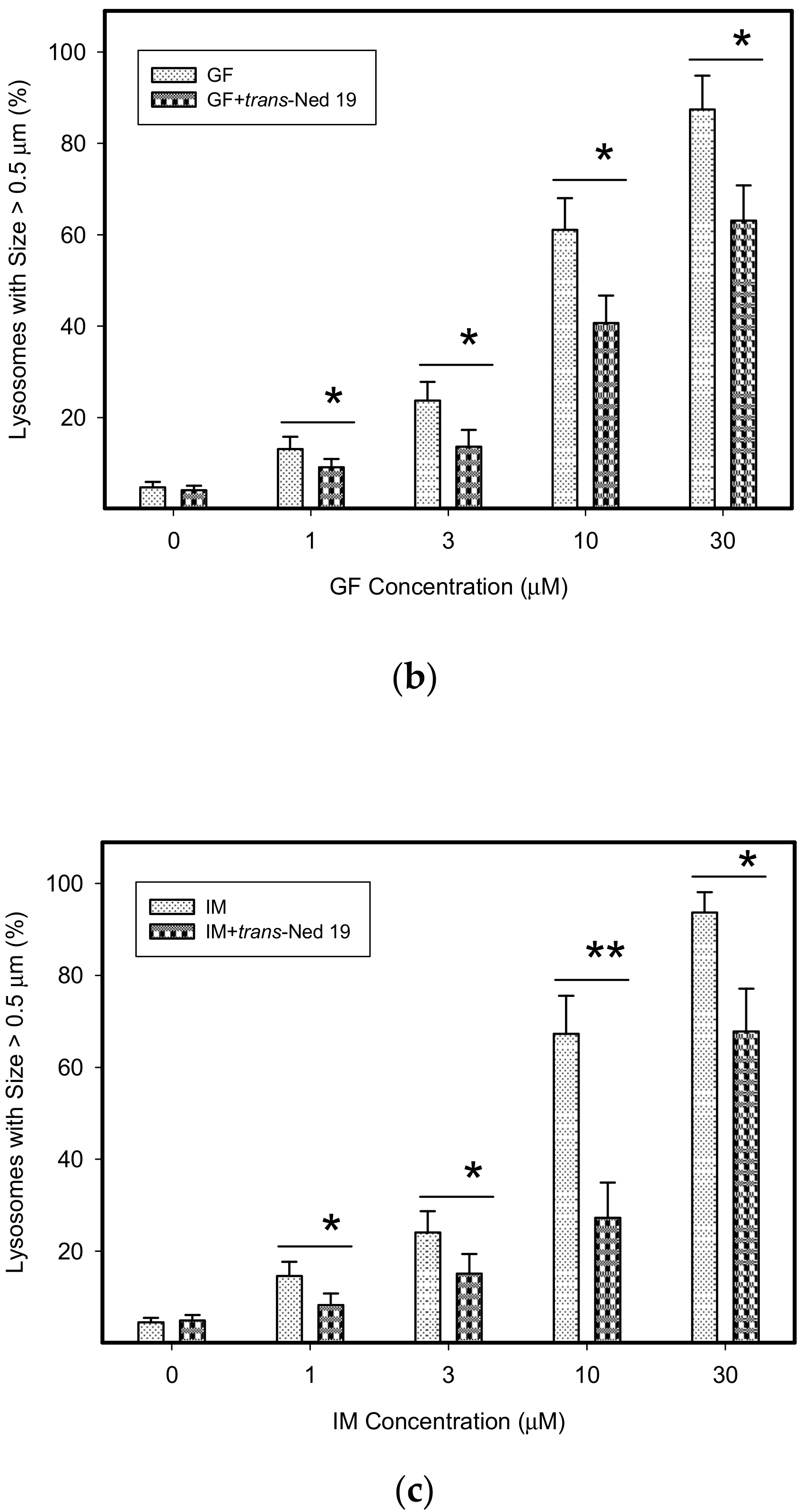
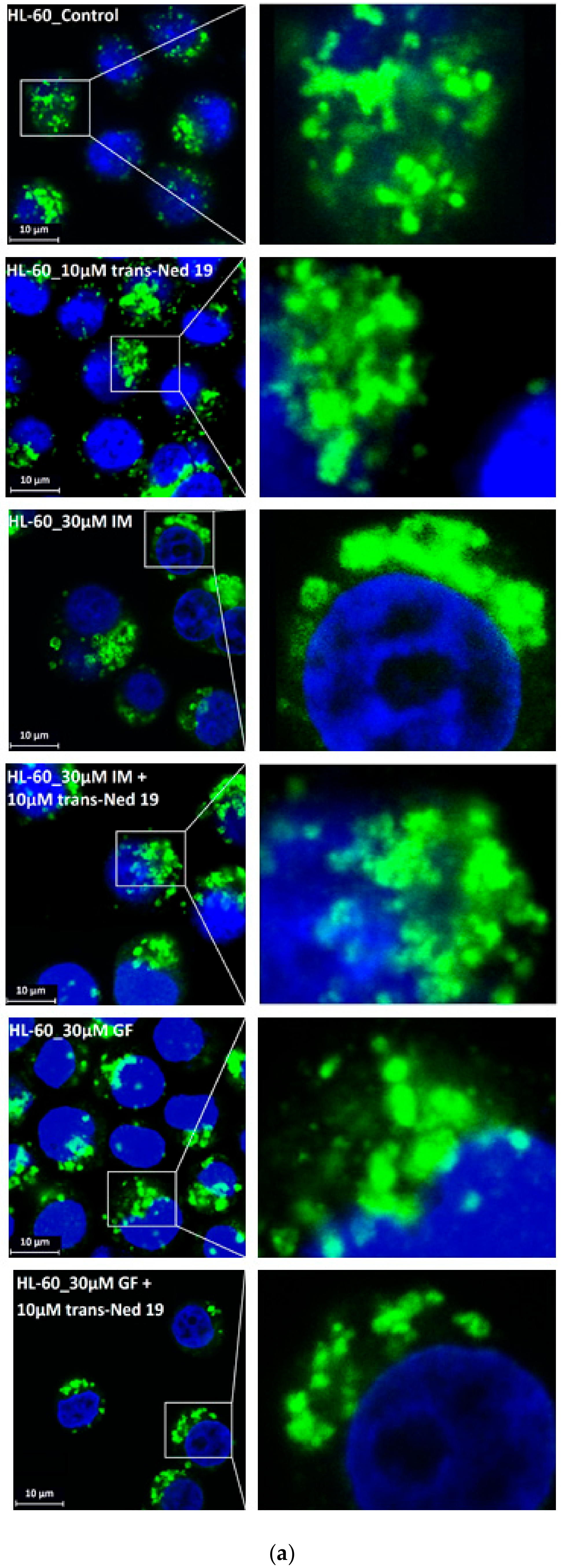
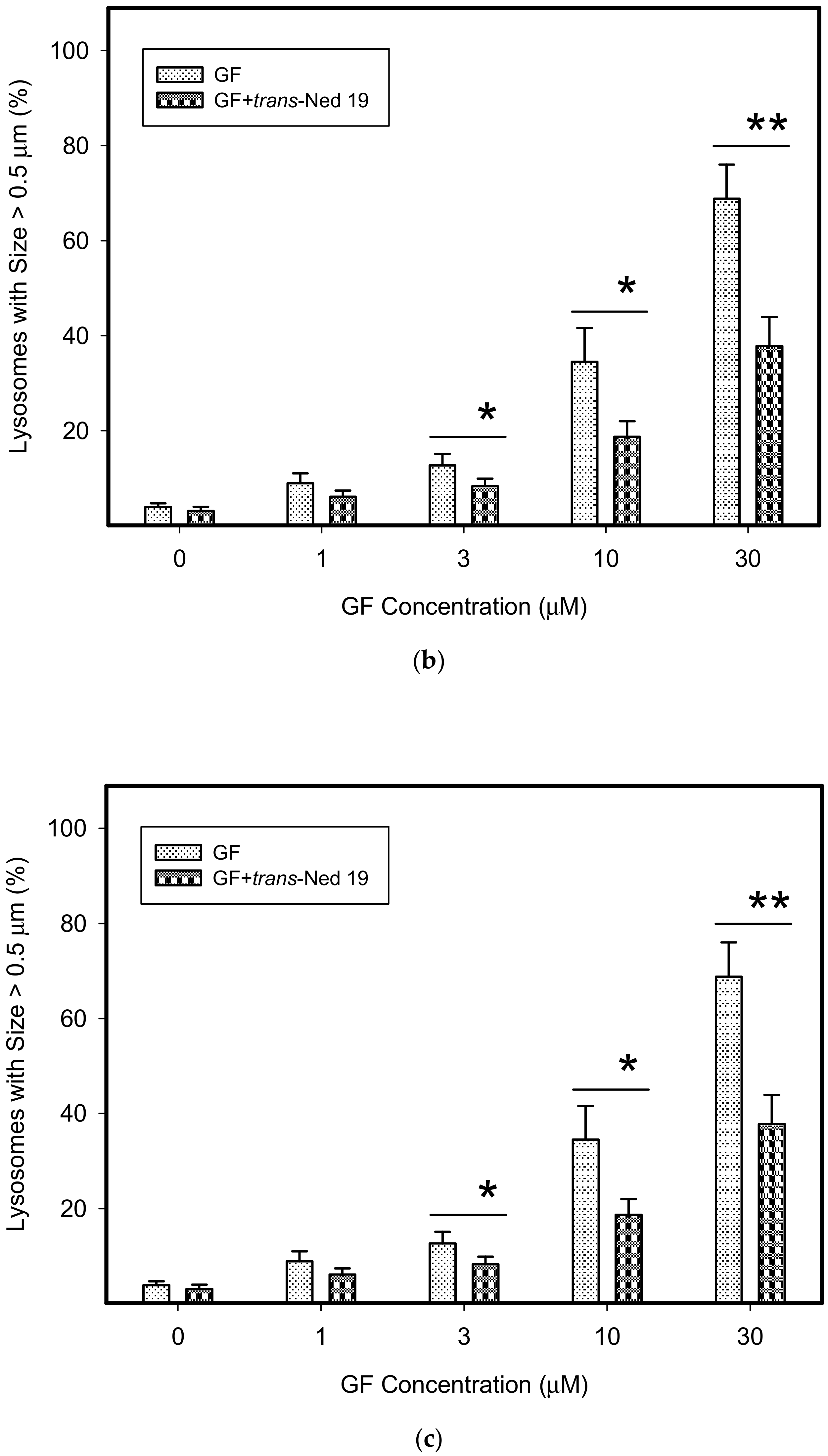
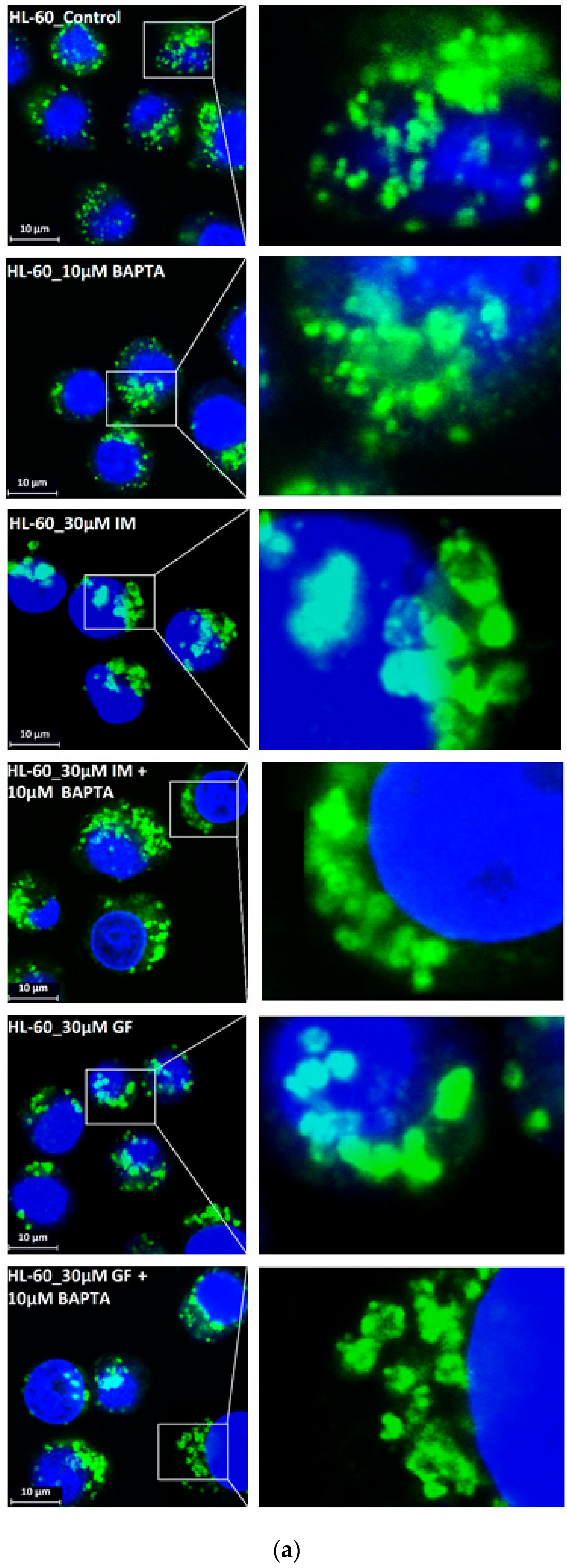
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Appelmans, F.; Wattiaux, R.; De Duve, C. Tissue fractionation studies. 5. The association of acid phosphatase with a special class of cytoplasmic granules in rat liver. Biochem. J. 1955, 59, 438–445. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. Lysosomes, a new group of cytoplasmic particles. In Subcellular Particles; Hayashi, T., Ed.; The Ronald Press Co.: New York, NY, USA, 1959; pp. 128–159. [Google Scholar]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Lűllmann-Rauch, R. History and morphology of the lysosome. In Lysosomes; Saftig, P., Ed.; Springer: New York, NY, USA, 2005. [Google Scholar]
- Boya, P. Lysosomal function and dysfunction: Mechanism and disease. Antioxid. Redox Signal. 2012, 17, 766–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, R.M.; Zoncu, R. The lysosome as a regulatory hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inpanathan, S.; Botelho, R.J. The lysosome signaling platform: Adapting with the times. Front. Cell Dev. Biol. 2019, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Futerman, A.H.; van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol. Ther. 2000, 85, 217–229. [Google Scholar] [CrossRef]
- Duvvuri, M.; Krise, J.P. Intracellular drug sequestration events associated with the emergence of multidrug resistance: A mechanistic review. Front. Biosci. 2005, 10, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef]
- De Duve, C.; de Barsy, T.; Poole, B.; Trouet, A.; Tulkens, P.; van Hoof, F. Commentary. Lysosomotropic agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar] [CrossRef]
- MacIntyre, A.C.; Cutler, D.J. The potential role of lysosomes in tissue distribution of weak bases. Biopharm. Drug Dispos. 1988, 9, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, N.; Akiyama, S.; Kobayashi, M.; Kuwano, M. Lysosomotropic agents reverse multiple drug resistance in human cancer cells. Cancer Lett. 1986, 30, 251–259. [Google Scholar] [CrossRef]
- Hindenburg, A.A.; Gervasoni, J.E., Jr.; Krishna, S.; Stewart, V.J.; Rosado, M.; Lutzky, J.; Bhalla, K.; Baker, M.A.; Taub, R.N. Intracellular distribution and pharmacokinetics of daunorubicin in anthracycline-sensitive and -resistant HL-60 cells. Cancer Res. 1989, 49, 4607–4614. [Google Scholar]
- Gervasoni, J.E., Jr.; Fields, S.Z.; Krishna, S.; Baker, M.A.; Rosado, M.; Thuraisamy, K.; Hindenburg, A.A.; Taub, R.N. Subcellular distribution of daunorubicin in P-glycoprotein-positive and -negative drug-resistant cell lines using laser-assisted confocal microscopy. Cancer Res. 1991, 51, 4955–4963. [Google Scholar]
- Hurwitz, S.J.; Terashima, M.; Mizunuma, N.; Slapak, C.A. Vesicular anthracycline accumulation in doxorubicin-selected U-937 cells: Participation of lysosomes. Blood 1997, 89, 3745–3754. [Google Scholar] [CrossRef] [Green Version]
- Chapuy, B.; Panse, M.; Radunski, U.; Koch, R.; Wenzel, D.; Inagaki, N.; Haase, D.; Truemper, L.; Wulf, G.G. ABC transporter A3 facilitates lysosomal sequestration of imatinib and modulates susceptibility of chronic myeloid leukemia cell lines to this drug. Haematologica 2009, 94, 1528–1536. [Google Scholar] [CrossRef] [Green Version]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [Green Version]
- Colombo, F.; Trombetta, E.; Cetrangolo, P.; Maggioni, M.; Razini, P.; De Santis, F.; Torrente, Y.; Prati, D.; Torresani, E.; Porretti, L. Giant lysosomes as a chemotherapy resistance mechanism in hepatocellular carcinoma cells. PLoS ONE 2014, 9, e114787. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, S.; Cormerais, Y.; Dufies, M.; Grépin, R.; Colosetti, P.; Belaid, A.; Parola, J.; Martin, A.; Lacas-Gervais, S.; Mazure, N.M.; et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015, 11, 1891–1904. [Google Scholar] [CrossRef]
- Gotink, K.J.; Rovithi, M.; de Haas, R.R.; Honeywell, R.J.; Dekker, H.; Poel, D.; Azijli, K.; Peters, G.J.; Broxterman, H.J.; Verheul, H.M. Cross-resistance to clinically used tyrosine kinase inhibitors sunitinib, sorafenib and pazopanib. Cell. Oncol. 2015, 38, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Mlejnek, P.; Novak, O.; Dolezel, P. A non-radioactive assay for precise determination of intracellular levels of imatinib and its main metabolite in Bcr-Abl positive cells. Talanta 2011, 83, 1466–1471. [Google Scholar] [CrossRef]
- Krumpochova, P.; Kocurova, A.; Dolezel, P.; Mlejnek, P. Assay for determination of daunorubicin in cancer cells with multidrug resistance phenotype. J. Chromatogr. B 2011, 879, 1875–1880. [Google Scholar] [CrossRef]
- Kosztyu, P.; Bukvova, R.; Dolezel, P.; Mlejnek, P. Resistance to daunorubicin, imatinib, or nilotinib depends on expression levels of ABCB1 and ABCG2 in human leukemia cells. Chem. Biol. Interact. 2014, 219, 203–210. [Google Scholar] [CrossRef]
- Burger, H.; den Dekker, A.T.; Segeletz, S.; Boersma, A.W.; de Bruijn, P.; Debiec-Rychter, M.; Taguchi, T.; Sleijfer, S.; Sparreboom, A.; Mathijssen, R.H.; et al. Lysosomal sequestration determines intracellular imatinib levels. Mol. Pharmacol. 2015, 88, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Ruzickova, E.; Skoupa, N.; Dolezel, P.; Smith, D.A.; Mlejnek, P. The lysosomal sequestration of tyrosine kinase inhibitors and drug resistance. Biomolecules 2019, 9, 675. [Google Scholar] [CrossRef] [Green Version]
- Frydrych, I.; Mlejnek, P. Serine protease inhibitors N-alpha-tosyl-L-lysinyl-chloromethylketone (TLCK) and N-tosyl-L-phenylalaninyl-chloromethylketone (TPCK) do not inhibit caspase-3 and caspase-7 processing in cells exposed to pro-apoptotic inducing stimuli. J. Cell. Biochem. 2008, 105, 1501–1506. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Mlejnek, P.; Kosztyu, P.; Dolezel, P.; Kimura, Y.; Cizkova, K.; Ruzickova, E. Estimation of ABCB1 concentration in plasma membrane. J. Cell. Biochem. 2019, 120, 18406–18414. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Kantarjian, H.; Alattar, M.L.; Jabbour, E.; Sasaki, K.; Nogueras Gonzalez, G.; Dellasala, S.; Pierce, S.; Verstovsek, S.; Wierda, W.; et al. Long-term molecular and cytogenetic response and survival outcomes with imatinib 400 mg, imatinib 800 mg, dasatinib, and nilotinib in patients with chronic-phase chronic myeloid leukaemia: Retrospective analysis of patient data from five clinical trials. Lancet Haematol. 2015, 2, e118–e128. [Google Scholar] [CrossRef] [Green Version]
- Cioccio, J.; Claxton, D. Therapy of acute myeloid leukemia: Therapeutic targeting of tyrosine kinases. Expert Opin. Investig. Drugs 2019, 28, 337–349. [Google Scholar] [CrossRef]
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12, e0173771. [Google Scholar] [CrossRef] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Pryor, P.R.; Mullock, B.M.; Bright, N.A.; Gray, S.R.; Luzio, J.P. The role of intraorganellar Ca(2+) in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J. Cell Biol. 2000, 149, 1053–1062. [Google Scholar] [CrossRef] [Green Version]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Galione, A. NAADP Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035071. [Google Scholar] [CrossRef] [PubMed]





























© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoupa, N.; Dolezel, P.; Mlejnek, P. Lysosomal Fusion: An Efficient Mechanism Increasing Their Sequestration Capacity for Weak Base Drugs without Apparent Lysosomal Biogenesis. Biomolecules 2020, 10, 77. https://doi.org/10.3390/biom10010077
Skoupa N, Dolezel P, Mlejnek P. Lysosomal Fusion: An Efficient Mechanism Increasing Their Sequestration Capacity for Weak Base Drugs without Apparent Lysosomal Biogenesis. Biomolecules. 2020; 10(1):77. https://doi.org/10.3390/biom10010077
Chicago/Turabian StyleSkoupa, Nikola, Petr Dolezel, and Petr Mlejnek. 2020. "Lysosomal Fusion: An Efficient Mechanism Increasing Their Sequestration Capacity for Weak Base Drugs without Apparent Lysosomal Biogenesis" Biomolecules 10, no. 1: 77. https://doi.org/10.3390/biom10010077
APA StyleSkoupa, N., Dolezel, P., & Mlejnek, P. (2020). Lysosomal Fusion: An Efficient Mechanism Increasing Their Sequestration Capacity for Weak Base Drugs without Apparent Lysosomal Biogenesis. Biomolecules, 10(1), 77. https://doi.org/10.3390/biom10010077