Comparative Assessment of Intrinsic Disorder Predictions with a Focus on Protein and Nucleic Acid-Binding Proteins



Abstract
:1. Introduction
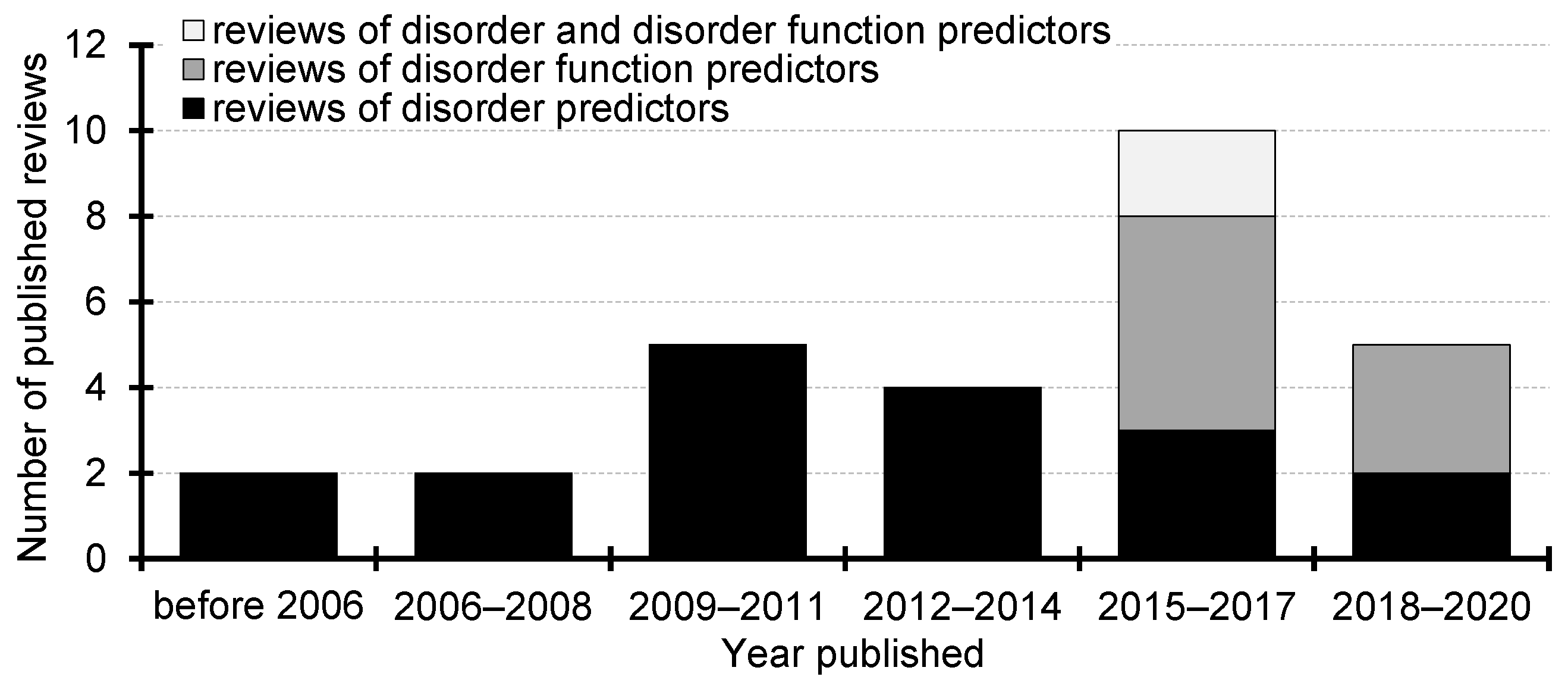
2. Surveys of the Intrinsic Disorder Predictors
3. Setup for Comparative Analysis
3.1. Selection of Disorder Predictors
3.2. Benchmark Dataset
3.3. Assessment of Predictive Performance
4. Comparative Assessments
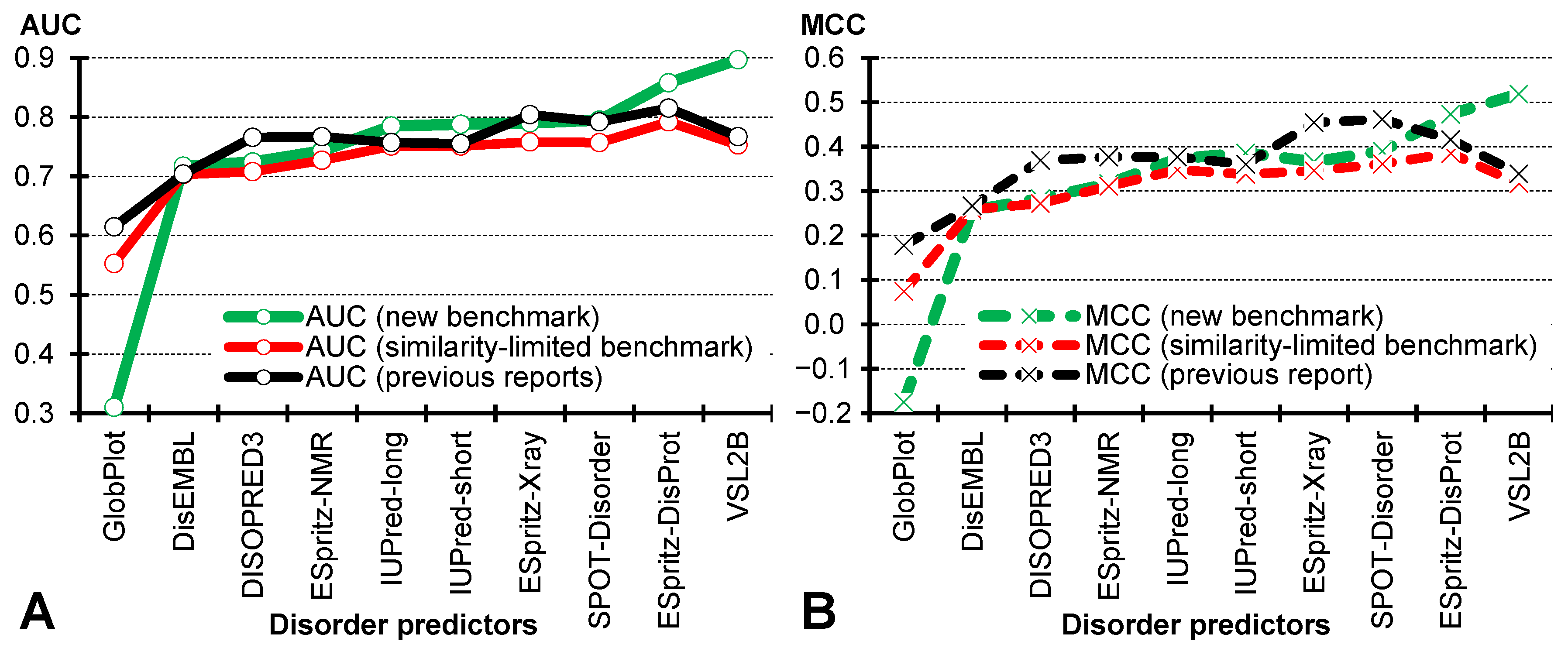
4.1. Impact of Sequence Similarity and Experimental Validation of Annotations
4.2. Comparative Assessment on the Benchmark Dataset
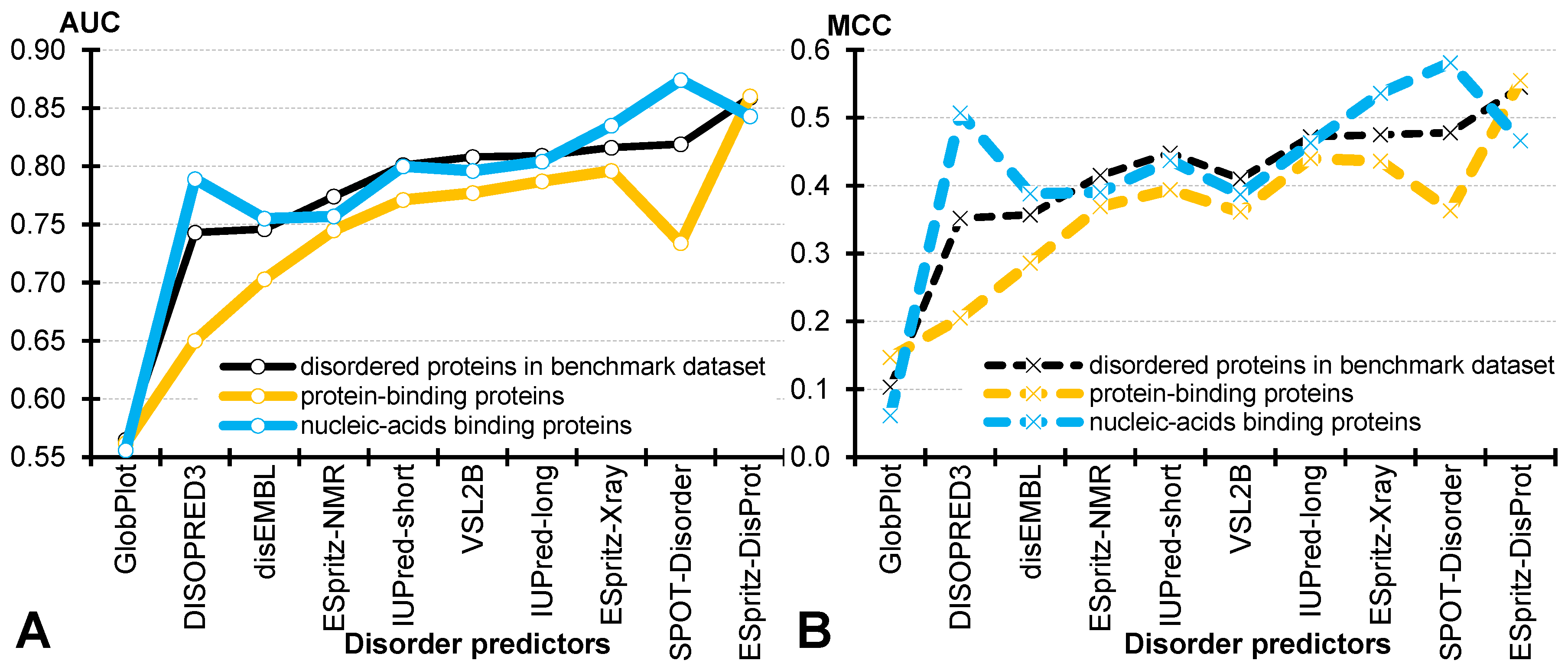
4.3. Comparative Assessment for the Disordered Protein-Binding and Nucleic Acid-Binding Proteins
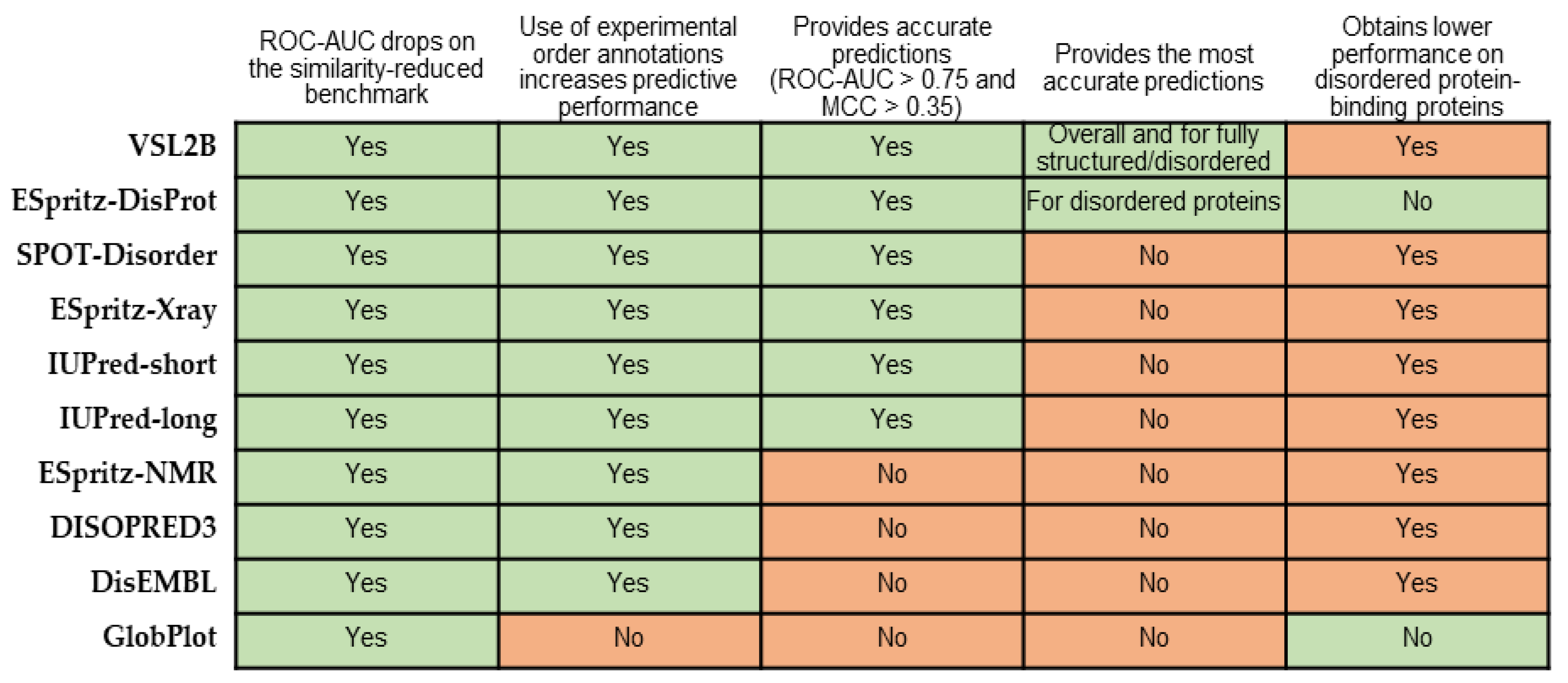
5. Summary
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lieutaud, P.; Ferron, F.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. How disordered is my protein and what is its disorder for? A guide through the “dark side” of the protein universe. Intrinsically Disord. Proteins 2016, 4, e1259708. [Google Scholar] [CrossRef] [Green Version]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Uversky, V.N.; Dunker, A.K.; Kurgan, L. Introduction to intrinsically disordered proteins and regions. In Intrinsically Disordered Proteins; Salvi, N., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 1–34. [Google Scholar] [CrossRef]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Intrinsic Disorder, Protein-Protein Interactions, and Disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Peng, Z.; Mizianty, M.J.; Kurgan, L. Genome-scale prediction of proteins with long intrinsically disordered regions. Proteins 2014, 82, 145–158. [Google Scholar] [CrossRef]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, A.; Brookes, D.H.; Yost, S.R.; Dyson, H.J.; Forman-Kay, J.D.; Gunter, D.; Head-Gordon, M.; Hura, G.L.; Pande, V.S.; Wemmer, D.E.; et al. Finding Our Way in the Dark Proteome. J. Am. Chem. Soc. 2016, 138, 9730–9742. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Wang, K.; Song, J.; Uversky, V.N.; Kurgan, L. Taxonomic Landscape of the Dark Proteomes: Whole-Proteome Scale Interplay Between Structural Darkness, Intrinsic Disorder, and Crystallization Propensity. Proteomics 2018, 18, e1800243. [Google Scholar] [CrossRef]
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins: The Dark Horse of the Dark Proteome. Proteomics 2018, 18, 1800061. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Oldfield, C.J.; Xue, B.; Mizianty, M.J.; Dunker, A.K.; Kurgan, L.; Uversky, V.N. A creature with a hundred waggly tails: Intrinsically disordered proteins in the ribosome. Cell. Mol. Life Sci. 2014, 71, 1477–1504. [Google Scholar] [CrossRef]
- Peng, Z.; Mizianty, M.J.; Xue, B.; Kurgan, L.; Uversky, V.N. More than just tails: Intrinsic disorder in histone proteins. Mol. Biosyst. 2012, 8, 1886–1901. [Google Scholar] [CrossRef]
- Wang, C.; Uversky, V.N.; Kurgan, L. Disordered nucleiome: Abundance of intrinsic disorder in the DNA- and RNA-binding proteins in 1121 species from Eukaryota, Bacteria and Archaea. Proteomics 2016, 16, 1486–1498. [Google Scholar] [CrossRef]
- Meng, F.; Na, I.; Kurgan, L.; Uversky, V.N. Compartmentalization and Functionality of Nuclear Disorder: Intrinsic Disorder and Protein-Protein Interactions in Intra-Nuclear Compartments. Int. J. Mol. Sci. 2015, 17, 24. [Google Scholar] [CrossRef] [Green Version]
- Fuxreiter, M.; Toth-Petroczy, A.; Kraut, D.A.; Matouschek, A.; Lim, R.Y.; Xue, B.; Kurgan, L.; Uversky, V.N. Disordered proteinaceous machines. Chem. Rev. 2014, 114, 6806–6843. [Google Scholar] [CrossRef] [Green Version]
- Na, I.; Meng, F.; Kurgan, L.; Uversky, V.N. Autophagy-related intrinsically disordered proteins in intra-nuclear compartments. Mol. Biosyst. 2016, 12, 2798–2817. [Google Scholar] [CrossRef]
- Peng, Z.; Xue, B.; Kurgan, L.; Uversky, V.N. Resilience of death: Intrinsic disorder in proteins involved in the programmed cell death. Cell Death Differ. 2013, 20, 1257–1267. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Uversky, V.N. Intrinsic disorder in proteins involved in the innate antiviral immunity: Another flexible side of a molecular arms race. J. Mol. Biol. 2014, 426, 1322–1350. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Kragelund, B.B. Functions of intrinsic disorder in transmembrane proteins. Cell. Mol. Life Sci. 2017, 74, 3205–3224. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Xue, B.; Dolan, P.T.; LaCount, D.J.; Kurgan, L.; Uversky, V.N. The intrinsic disorder status of the human hepatitis C virus proteome. Mol. Biosyst. 2014, 10, 1345–1363. [Google Scholar] [CrossRef]
- Yan, J.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Molecular recognition features (MoRFs) in three domains of life. Mol. Biosyst. 2016, 12, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef]
- Receveur-Brechot, V.; Bourhis, J.M.; Uversky, V.N.; Canard, B.; Longhi, S. Assessing protein disorder and induced folding. Proteins 2006, 62, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic Disorder-based Protein Interactions and their Modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhivker, G.M.; Bouzida, D.; Gehlhaar, D.K.; Rejto, P.A.; Freer, S.T.; Rose, P.W. Simulating disorder-order transitions in molecular recognition of unstructured proteins: Where folding meets binding. Proc. Natl. Acad. Sci. USA 2003, 100, 5148–5153. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Hu, G.; Yang, J.; Peng, Z.; Uversky, V.N.; Kurgan, L. In various protein complexes, disordered protomers have large per-residue surface areas and area of protein-, DNA- and RNA-binding interfaces. FEBS Lett. 2015, 589, 2561–2569. [Google Scholar] [CrossRef]
- Varadi, M.; Zsolyomi, F.; Guharoy, M.; Tompa, P. Functional Advantages of Conserved Intrinsic Disorder in RNA-Binding Proteins. PLoS ONE 2015, 10, e0139731. [Google Scholar] [CrossRef]
- Dyson, H.J. Roles of intrinsic disorder in protein-nucleic acid interactions. Mol. Biosyst. 2012, 8, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9 (Suppl. S1), S1. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Hatos, A.; Hajdu-Soltesz, B.; Monzon, A.M.; Palopoli, N.; Alvarez, L.; Aykac-Fas, B.; Bassot, C.; Benitez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, D.; Tabaro, F.; Paladin, L.; Necci, M.; Micetic, I.; Camilloni, C.; Davey, N.; Dosztanyi, Z.; Meszaros, B.; Monzon, A.M.; et al. MobiDB 3.0: More annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018, 46, D471–D476. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, T.; Romero, P.R.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in the Protein Data Bank. J. Biomol. Struct. Dyn. 2007, 24, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Amemiya, T.; Sakamoto, S.; Nobe, Y.; Hosoda, K.; Kado, Y.; Murakami, S.D.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014, 42, D320–D325. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Giollo, M.; Di Domenico, T.; Ferrari, C.; Zimmermann, O.; Tosatto, S.C. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015, 31, 201–208. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090. [Google Scholar] [CrossRef]
- Meng, F.; Uversky, V.; Kurgan, L. Computational Prediction of Intrinsic Disorder in Proteins. Curr. Protoc. Protein Sci. 2017, 88, 2.16.11–2.16.14. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Liu, B. A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief. Bioinform. 2019, 20, 330–346. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Vullo, A.; Pollastri, G.; Tosatto, S.C.E. CSpritz: Accurate prediction of protein disorder segments with annotation for homology, secondary structure and linear motifs. Nucleic Acids Res. 2011, 39, W190–W196. [Google Scholar] [CrossRef]
- Fan, X.; Kurgan, L. Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J. Biomol. Struct. Dyn. 2014, 32, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein Disorder Prediction: Implications for Structural Proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.J.; Acton, T.B.; Montelione, G.T. DisMeta: A meta server for construct design and optimization. Methods Mol. Biol. 2014, 1091, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Ward, J.J. Prediction of disordered regions in proteins from position specific score matrices. Proteins Struct. Funct. Bioinform. 2003, 53, 573–578. [Google Scholar] [CrossRef]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Cheng, J.; Sweredoski, M.; Baldi, P. Accurate Prediction of Protein Disordered Regions by Mining Protein Structure Data. Data Min. Knowl. Disc. 2005, 11, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Hecker, J.; Yang, J.Y.; Cheng, J. Protein disorder prediction at multiple levels of sensitivity and specificity. BMC Genom. 2008, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [Green Version]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [Green Version]
- Dosztányi, Z.; Csizmók, V.; Tompa, P.; Simon, I. The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Schlessinger, A.; Punta, M.; Yachdav, G.; Kajan, L.; Rost, B. Improved disorder prediction by combination of orthogonal approaches. PLoS ONE 2009, 4, e4433. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, L.P.; Bujnicki, J.M. MetaDisorder: A meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinform. 2012, 13, 111. [Google Scholar] [CrossRef] [Green Version]
- Mizianty, M.J.; Stach, W.; Chen, K.; Kedarisetti, K.D.; Disfani, F.M.; Kurgan, L. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010, 26, i489–i496. [Google Scholar] [CrossRef] [Green Version]
- Mizianty, M.J.; Uversky, V.; Kurgan, L. Prediction of intrinsic disorder in proteins using MFDp2. Methods Mol. Biol. 2014, 1137, 147–162. [Google Scholar] [CrossRef]
- Mizianty, M.J.; Peng, Z.; Kurgan, L. MFDp2-Accurate predictor of disorder in proteins by fusion of disorder probabilities, content and profiles. Intrinsically Disord. Proteins 2013, 1, e24428. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Dosztanyi, Z.; Tosatto, S.C.E. MobiDB-lite: Fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics 2017, 33, 1402–1404. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef]
- Deng, X.; Eickholt, J.; Cheng, J. PreDisorder: Ab initio sequence-based prediction of protein disordered regions. BMC Bioinform. 2009, 10, 436. [Google Scholar] [CrossRef] [Green Version]
- Hanson, J.; Yang, Y.; Paliwal, K.; Zhou, Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics 2017, 33, 685–692. [Google Scholar] [CrossRef] [Green Version]
- Hanson, J.; Paliwal, K.K.; Litfin, T.; Zhou, Y. SPOT-Disorder2: Improved Protein Intrinsic Disorder Prediction by Ensembled Deep Learning. Genom. Proteom. Bioinform. 2020, 17, 645–656. [Google Scholar] [CrossRef]
- Hanson, J.; Paliwal, K.K.; Zhou, Y. Accurate Single-Sequence Prediction of Protein Intrinsic Disorder by an Ensemble of Deep Recurrent and Convolutional Architectures. J. Chem. Inf. Model. 2018, 58, 2369–2376. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 2005, 61 (Suppl. S7), 176–182. [Google Scholar] [CrossRef]
- Barik, A.; Katuwawala, A.; Hanson, J.; Paliwal, K.; Zhou, Y.; Kurgan, L. DEPICTER: Intrinsic Disorder and Disorder Function Prediction Server. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Meng, F.; Kurgan, L. DFLpred: High-throughput prediction of disordered flexible linker regions in protein sequences. Bioinformatics 2016, 32, i341–i350. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Kurgan, L. High-throughput prediction of disordered moonlighting regions in protein sequences. Proteins 2018. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, C.; Uversky, V.N.; Kurgan, L. Prediction of Disordered RNA, DNA, and Protein Binding Regions Using DisoRDPbind. Methods Mol. Biol. 2017, 1484, 187–203. [Google Scholar] [CrossRef]
- Peng, Z.; Kurgan, L. High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015, 43, e121. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.L.; Kurgan, L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012, 13, 6–18. [Google Scholar] [CrossRef] [Green Version]
- Monastyrskyy, B.; Kryshtafovych, A.; Moult, J.; Tramontano, A.; Fidelis, K. Assessment of protein disorder region predictions in CASP10. Proteins 2014, 82 (Suppl. S2), 127–137. [Google Scholar] [CrossRef] [Green Version]
- Monastyrskyy, B.; Fidelis, K.; Moult, J.; Tramontano, A.; Kryshtafovych, A. Evaluation of disorder predictions in CASP9. Proteins 2011, 79 (Suppl. S10), 107–118. [Google Scholar] [CrossRef] [Green Version]
- Noivirt-Brik, O.; Prilusky, J.; Sussman, J.L. Assessment of disorder predictions in CASP8. Proteins 2009, 77 (Suppl. S9), 210–216. [Google Scholar] [CrossRef]
- Bordoli, L.; Kiefer, F.; Schwede, T. Assessment of disorder predictions in CASP7. Proteins 2007, 69 (Suppl. S8), 129–136. [Google Scholar] [CrossRef]
- Jin, Y.; Dunbrack, R.L., Jr. Assessment of disorder predictions in CASP6. Proteins 2005, 61 (Suppl. S7), 167–175. [Google Scholar] [CrossRef]
- Melamud, E.; Moult, J. Evaluation of disorder predictions in CASP5. Proteins 2003, 53 (Suppl. S6), 561–565. [Google Scholar] [CrossRef] [Green Version]
- Necci, M.; Piovesan, D.; Dosztanyi, Z.; Tompa, P.; Tosatto, S.C.E. A comprehensive assessment of long intrinsic protein disorder from the DisProt database. Bioinformatics 2018, 34, 445–452. [Google Scholar] [CrossRef]
- Pryor, E.E., Jr.; Wiener, M.C. A critical evaluation of in silico methods for detection of membrane protein intrinsic disorder. Biophys. J. 2014, 106, 1638–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katuwawala, A.; Oldfield, C.J.; Kurgan, L. Accuracy of protein-level disorder predictions. Brief. Bioinform. 2020, 21, 1509–1522. [Google Scholar] [CrossRef]
- Katuwawala, A.; Ghadermarzi, S.; Kurgan, L. Computational prediction of functions of intrinsically disordered regions. Prog. Mol. Biol. Transl. Sci. 2019, 166, 341–369. [Google Scholar] [CrossRef]
- Katuwawala, A.; Peng, Z.; Yang, J.; Kurgan, L. Computational Prediction of MoRFs, Short Disorder-to-order Transitioning Protein Binding Regions. Comput. Struct. Biotechnol. J. 2019, 17, 454–462. [Google Scholar] [CrossRef]
- Uversky, V.N. How to Predict Disorder in a Protein of Interest. Methods Mol. Biol. 2017, 1484, 137–158. [Google Scholar] [CrossRef]
- Li, J.; Feng, Y.; Wang, X.; Li, J.; Liu, W.; Rong, L.; Bao, J. An Overview of Predictors for Intrinsically Disordered Proteins over 2010–2014. Int. J. Mol. Sci. 2015, 16, 23446–23462. [Google Scholar] [CrossRef]
- Deng, X.; Gumm, J.; Karki, S.; Eickholt, J.; Cheng, J. An Overview of Practical Applications of Protein Disorder Prediction and Drive for Faster, More Accurate Predictions. Int. J. Mol. Sci. 2015, 16, 15384–15404. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, P.; Guharoy, M.; Tompa, P. Bioinformatics Approaches for Predicting Disordered Protein Motifs. Adv. Exp. Med. Biol. 2015, 870, 291–318. [Google Scholar] [CrossRef]
- Varadi, M.; Vranken, W.; Guharoy, M.; Tompa, P. Computational approaches for inferring the functions of intrinsically disordered proteins. Front. Mol. BioSci. 2015, 2, 45. [Google Scholar] [CrossRef] [Green Version]
- Atkins, J.D.; Boateng, S.Y.; Sorensen, T.; McGuffin, L.J. Disorder Prediction Methods, Their Applicability to Different Protein Targets and Their Usefulness for Guiding Experimental Studies. Int. J. Mol. Sci. 2015, 16, 19040–19054. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Eickholt, J.; Cheng, J. A comprehensive overview of computational protein disorder prediction methods. Mol. Biosyst. 2012, 8, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Orosz, F.; Ovadi, J. Proteins without 3D structure: Definition, detection and beyond. Bioinformatics 2011, 27, 1449–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. Bioinformatical approaches to characterize intrinsically disordered/unstructured proteins. Brief. Bioinform. 2010, 11, 225–243. [Google Scholar] [CrossRef]
- Uversky, V.N.; Radivojac, P.; Iakoucheva, L.M.; Obradovic, Z.; Dunker, A.K. Prediction of intrinsic disorder and its use in functional proteomics. Methods Mol. Biol. 2007, 408, 69–92. [Google Scholar] [CrossRef] [Green Version]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar]
- Piovesan, D.; Tabaro, F.; Micetic, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidovic, R.; Dosztanyi, Z.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2016, D1, D219–D227. [Google Scholar] [CrossRef] [Green Version]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The Database of Disordered Proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [Green Version]
- Necci, M.; Piovesan, D.; Tosatto, S.C.E. Critical Assessment of Protein Intrinsic Disorder Prediction. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, S.; Ma, J.Z.; Xu, J.B. AUCpreD: Proteome-level protein disorder prediction by AUC-maximized deep convolutional neural fields. Bioinformatics 2016, 32, 672–679. [Google Scholar] [CrossRef]
- Wang, S.; Weng, S.; Ma, J.; Tang, Q. DeepCNF-D: Predicting Protein Order/Disorder Regions by Weighted Deep Convolutional Neural Fields. Int. J. Mol. Sci. 2015, 16, 17315–17330. [Google Scholar] [CrossRef]
- Kedarisetti, K.D.; Mizianty, M.J.; Dick, S.; Kurgan, L. Improved sequence-based prediction of strand residues. J. Bioinform. Comput. Biol. 2011, 9, 67–89. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Tompa, P. Bioinformatics Approaches to the Structure and Function of Intrinsically Disordered Proteins. In From Protein Structure to Function with Bioinformatics; Rigden, J.D., Ed.; Springer: Dordrecht, The Netherlands, 2017; pp. 167–203. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Kurgan, L. Sequence Similarity Searching. Curr. Protoc. Protein Sci. 2019, 95, e71. [Google Scholar] [CrossRef] [Green Version]
- Dana, J.M.; Gutmanas, A.; Tyagi, N.; Qi, G.; O’Donovan, C.; Martin, M.; Velankar, S. SIFTS: Updated Structure Integration with Function, Taxonomy and Sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic Acids Res. 2019, 47, D482–D489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, Z.; Kurgan, L. Comprehensive review and empirical analysis of hallmarks of DNA-, RNA- and protein-binding residues in protein chains. Brief. Bioinform. 2019, 20, 1250–1268. [Google Scholar] [CrossRef]
- Zhang, J.; Ghadermarzi, S.; Kurgan, L. Prediction of protein-binding residues: Dichotomy of sequence-based methods developed using structured complexes vs. disordered proteins. Bioinformatics 2020, 36, 4729–4738. [Google Scholar] [CrossRef]
- Barik, A.; Kurgan, L. A comprehensive overview of sequence-based protein-binding residue predictions for structured and disordered regions. Protein Interact. 2020, 33–58. [Google Scholar] [CrossRef]
- Natole, M.; Ying, Y.; Lyu, S. Stochastic AUC Optimization Algorithms With Linear Convergence. Front. Appl. Math. Stat. 2019, 5, 30. [Google Scholar] [CrossRef]
- Hu, G.; Wu, Z.; Oldfield, C.; Wang, C.; Kurgan, L. Quality Assessment for the Putative Intrinsic Disorder in Proteins. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Hu, G.; Wang, K.; Kurgan, L. Exploratory Analysis of Quality Assessment of Putative Intrinsic Disorder in Proteins. In Proceedings of the 6th International Conference on Artificial Intelligence and Soft Computing, Zakopane, Poland, 27 May 2017; Volume 10245, pp. 722–732. [Google Scholar]
- Peng, Z.; Kurgan, L. On the complementarity of the consensus-based disorder prediction. Pac. Symp. BioComput. 2012, 2012, 176–187. [Google Scholar]
- Katuwawala, A.; Oldfield, C.; Kurgan, L. DISOselect: Disorder predictor selection at the protein level. Protein Sci. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Article | Target of Assessment | Suggested Best Disorder Predictors (Year Published) | Year Assessment Published | Year Most Recent Assessed Predictor Published | Number of Citations | Benchmark Dataset has Reduced Similarity with Training Sets of the Assessed Predictors |
|---|---|---|---|---|---|---|
| This article | disordered proteins; disordered protein-binding protein; disordered nucleic acids-binding proteins | N/A | N/A | 2018 | N/A | yes |
| [93] | disordered proteins | SPOT-Disorder (2017), DISOPRED3 (2015) | 2019 | 2017 | 4 | no |
| [91] | disordered proteins | Espritz (2012) | 2018 | 2017 | 33 | no |
| [45] | disordered proteins | DisEMBL (2003), IUPred (2005) | 2015 | 2012 | 121 | no |
| [85] | disordered proteins | DISOPRED3 (2015), PrDOS (2007), MFDp (2010) | 2014 | 2015 | 128 | no |
| [92] | disordered integral membrane proteins | PreDisorder (2009) | 2014 | 2012 | 12 | no |
| [84] | disordered proteins | MFDp (2010), MD (2009), PONDR-FIT (2010) | 2012 | 2010 | 149 | no |
| [86] | disordered proteins | PrDOS (2007), DISOPRED (2004) | 2011 | 2010 | 118 | no |
| [87] | disordered proteins | GS-MetaServer (2012), PreDisorder (2009) | 2009 | 2008 | 131 | no |
| [88] | disordered proteins | DISOPRED (2004), DISpro (2005) | 2007 | 2006 | 109 | no |
| [89] | disordered proteins | predictor by Obradovic et al. | 2005 | 2004 | 114 | no |
| [90] | disordered proteins | N/A | 2003 | 2002 | 97 | no |
| Dataset Characteristic | Complete Dataset | Protein-Binding Proteins | Nucleic Acids-Binding Proteins |
|---|---|---|---|
| Number of proteins | 357 | 108 | 15 |
| Number of residues | 186,337 | 38,221 | 5934 |
| Number of disordered residues | 31,608 | 14,125 | 1567 |
| Disorder content (% of disordered residues) | 0.17 | 0.37 | 0.26 |
| Predictor | Benchmark Dataset | Fully Disordered Proteins | Fully Ordered Proteins | Benchmark Dataset without Fully Ordered Proteins | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC | Precision | Sensitivity | FPR | MCC | Sensitivity | FPR | AUC | Precision | Sensitivity | FPR | MCC | |
| VSL2B | 0.897 | 0.609 | 0.845 | 0.204 | 0.519 | 0.925 | 0.000 | 0.805+ | 0.611 | 0.845 | 0.399 | 0.404 |
| ESpritz-DisProt | 0.858+ | 0.593 | 0.487 | 0.060 | 0.473 | 0.811 | 0.052 | 0.842 | 0.685 | 0.487 | 0.067 | 0.486 |
| SPOT-Disorder | 0.795+ | 0.334 | 0.756 | 0.261 | 0.390 | 0.662 | 0.290 | 0.826+ | 0.578 | 0.756 | 0.234 | 0.485 |
| ESpritz-Xray | 0.790+ | 0.375 | 0.623 | 0.193 | 0.366 | 0.702 | 0.226 | 0.812+ | 0.586 | 0.623 | 0.160 | 0.459 |
| IUPred-short | 0.788+ | 0.431 | 0.613 | 0.170 | 0.386 | 0.692 | 0.176 | 0.801+ | 0.607 | 0.613 | 0.165 | 0.444 |
| IUPred-long | 0.785+ | 0.422 | 0.693 | 0.233 | 0.373 | 0.834 | 0.262 | 0.806+ | 0.625 | 0.693 | 0.206 | 0.463 |
| ESpritz-NMR | 0.743+ | 0.336 | 0.721 | 0.310 | 0.317 | 0.774 | 0.351 | 0.776+ | 0.563 | 0.721 | 0.272 | 0.414 |
| DISOPRED3 | 0.724+ | 0.294 | 0.653 | 0.293 | 0.283 | 0.662 | 0.340 | 0.767+ | 0.513 | 0.653 | 0.248 | 0.380 |
| DisEMBL | 0.717+ | 0.308 | 0.439 | 0.162 | 0.257 | 0.559 | 0.193 | 0.741+ | 0.520 | 0.439 | 0.132 | 0.336 |
| GlobPlot | 0.310+ | 0.122 | 0.428 | 0.655 | −0.175 | 0.388 | 1.000 | 0.563+ | 0.332 | 0.428 | 0.326 | 0.096 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katuwawala, A.; Kurgan, L. Comparative Assessment of Intrinsic Disorder Predictions with a Focus on Protein and Nucleic Acid-Binding Proteins. Biomolecules 2020, 10, 1636. https://doi.org/10.3390/biom10121636
Katuwawala A, Kurgan L. Comparative Assessment of Intrinsic Disorder Predictions with a Focus on Protein and Nucleic Acid-Binding Proteins. Biomolecules. 2020; 10(12):1636. https://doi.org/10.3390/biom10121636
Chicago/Turabian StyleKatuwawala, Akila, and Lukasz Kurgan. 2020. "Comparative Assessment of Intrinsic Disorder Predictions with a Focus on Protein and Nucleic Acid-Binding Proteins" Biomolecules 10, no. 12: 1636. https://doi.org/10.3390/biom10121636
APA StyleKatuwawala, A., & Kurgan, L. (2020). Comparative Assessment of Intrinsic Disorder Predictions with a Focus on Protein and Nucleic Acid-Binding Proteins. Biomolecules, 10(12), 1636. https://doi.org/10.3390/biom10121636