Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant?



{kind=link}
{kind=link}
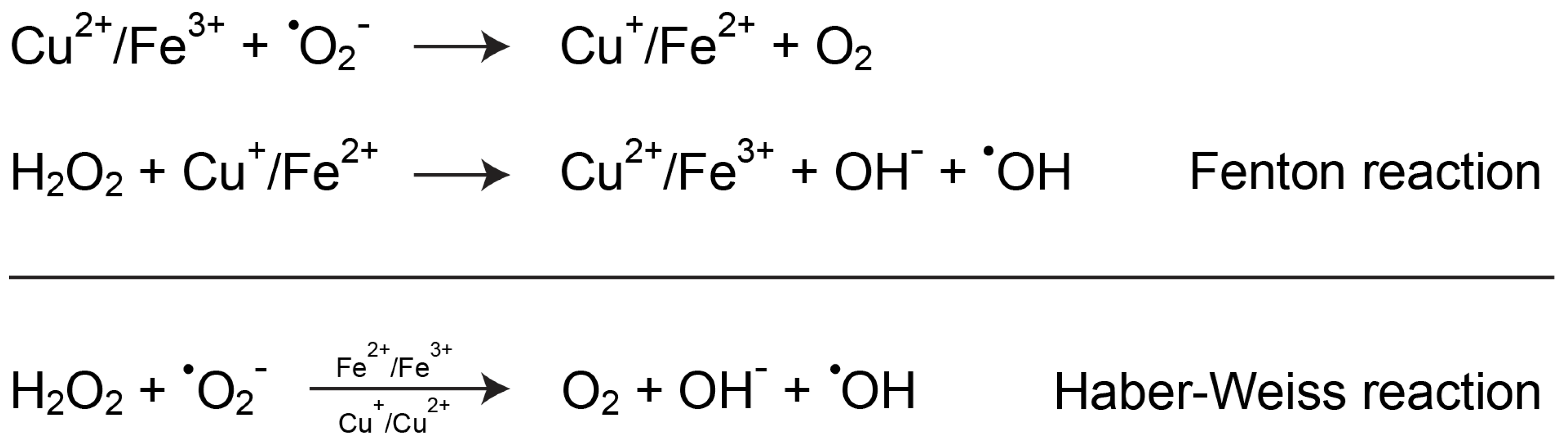{kind=link}
{kind=link}
Abstract
:1. Introduction
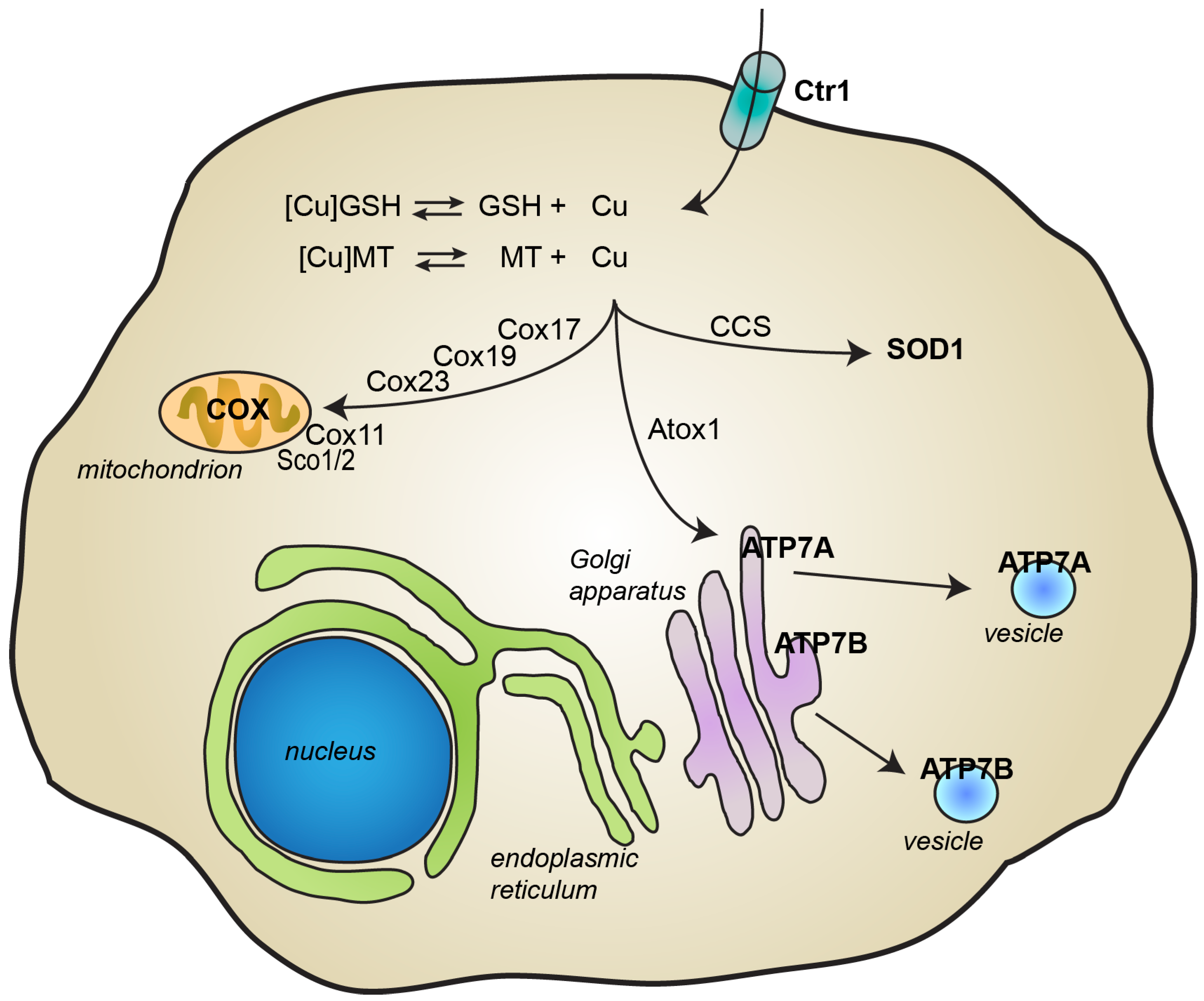
2. Copper Metabolism
3. Alteration of Copper Homeostasis and Pathological Consequences
4. Parkinson’s Disease
5. Mechanisms of Copper Toxicity in Parkinson’s Disease
5.1. Copper and Oxidative Stress
5.2. Copper and Dopamine Oxidation
5.3. Copper Effects on α-Synuclein Aggregation
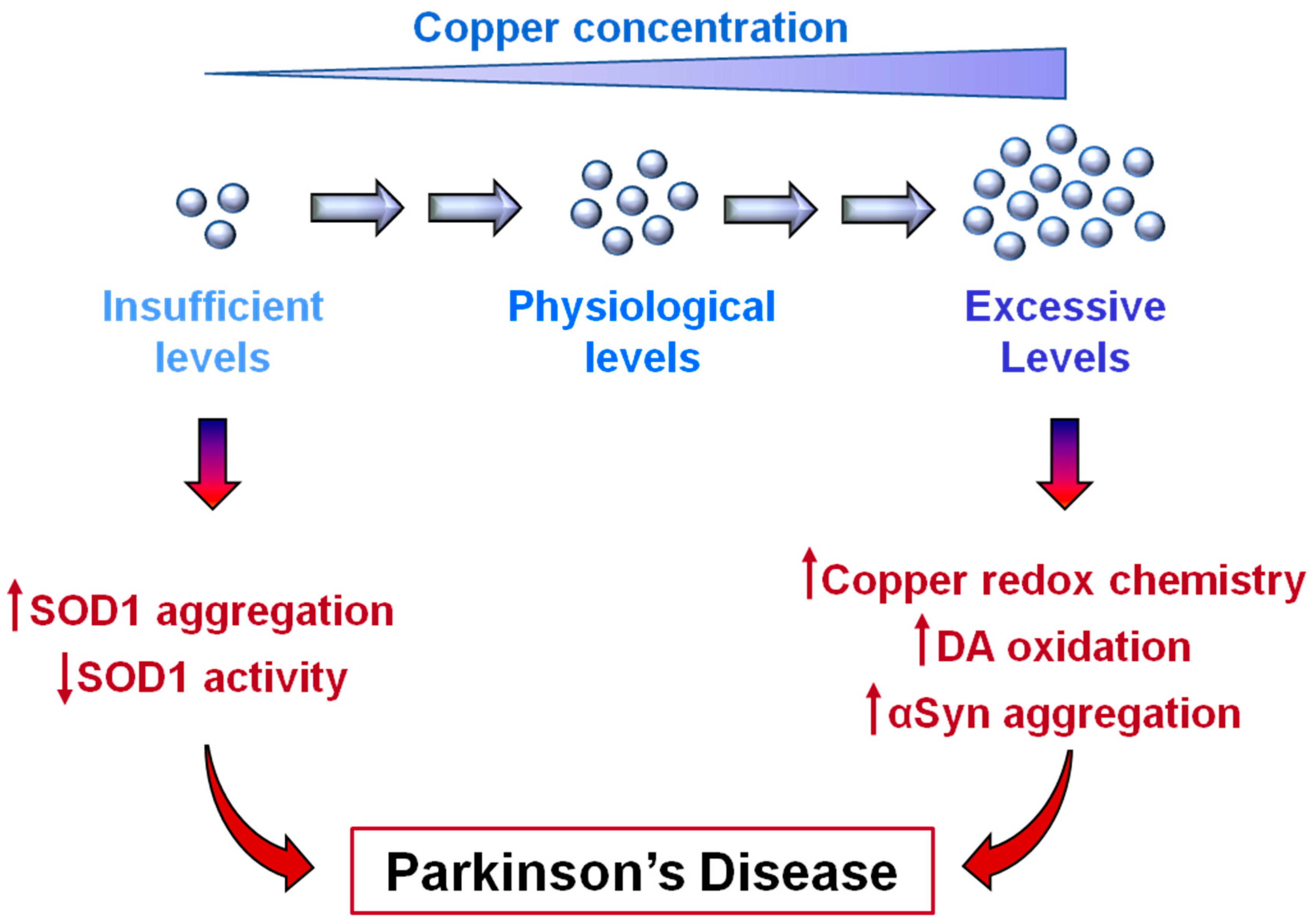
6. Copper Deficiency in Parkinson’s Disease
7. A New Proposed Role of SOD1 in Parkinson’s Disease Pathology
8. A therapeutic Conundrum
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Szerdahelyi, P.; Kasa, P. Histochemical demonstration of copper in normal rat brain and spinal cord. Evidence of localization in glial cells. Histochemistry 1986, 85, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Lewinska-Preis, L.; Jablonska, M.; Fabianska, M.J.; Kita, A. Bioelements and mineral matter in human livers from the highly industrialized region of the upper silesia coal basin (poland). Env. Geochem. Health 2011, 33, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Bulcke, F.; Dringen, R.; Scheiber, I.F. Neurotoxicity of copper. Adv. Neurobiol. 2017, 18, 313–343. [Google Scholar] [PubMed]
- Navarro, J.A.; Schneuwly, S. Copper and zinc homeostasis: Lessons from drosophila melanogaster. Front. Genet. 2017, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Copper and copper nanoparticles toxicity and their impact on basic functions in the body. Bratisl. Lek. Listy. 2019, 120, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Roeser, H.P.; Lee, G.R.; Nacht, S.; Cartwright, G.E. The role of ceruloplasmin in iron metabolism. J. Clin. Invest. 1970, 49, 2408–2417. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef]
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar] [CrossRef]
- Davies, K.M.; Mercer, J.F.; Chen, N.; Double, K.L. Copper dyshomoeostasis in parkinson’s disease: Implications for pathogenesis and indications for novel therapeutics. Clin. Sci. (Lond) 2016, 130, 565–574. [Google Scholar] [CrossRef]
- Ackerman, C.M.; Chang, C.J. Copper signaling in the brain and beyond. J. Biol. Chem. 2018, 293, 4628–4635. [Google Scholar] [CrossRef] [Green Version]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumaa, P. Copper chaperones. The concept of conformational control in the metabolism of copper. FEBS Lett. 2013, 587, 1902–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council (U.S.); Committee on Copper in Drinking Water. Copper in Drinking Water; National Academy Press: Washington, DC, USA, 2000; 147p. [Google Scholar]
- Tumer, Z.; Moller, L.B. Menkes disease. Eur. J. Hum. Genet. 2010, 18, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaler, S.G. Atp7a-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litwin, T.; Gromadzka, G.; Szpak, G.M.; Jablonka-Salach, K.; Bulska, E.; Czlonkowska, A. Brain metal accumulation in wilson’s disease. J. Neurol. Sci. 2013, 329, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113. [Google Scholar] [CrossRef] [Green Version]
- De Lazzari, F.; Bubacco, L.; Whitworth, A.J.; Bisaglia, M. Superoxide radical dismutation as new therapeutic strategy in parkinson’s disease. Aging Dis. 2018, 9, 716–728. [Google Scholar] [CrossRef] [Green Version]
- Youdim, M.B.; Ben-Shachar, D.; Riederer, P. Is parkinson’s disease a progressive siderosis of substantia nigra resulting in iron and melanin induced neurodegeneration? Acta Neurol. Scand. Suppl. 1989, 126, 47–54. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef]
- Perry, T.L.; Godin, D.V.; Hansen, S. Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci. Lett. 1982, 33, 305–310. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in parkinson’s disease. Ann. Neurol. 2003, 3 (Suppl. 3), S26–S36. [Google Scholar] [CrossRef] [PubMed]
- Zayed, J.; Ducic, S.; Campanella, G.; Panisset, J.C.; Andre, P.; Masson, H.; Roy, M. [Environmental factors in the etiology of parkinson’s disease]. Can. J. Neurol. Sci. 1990, 17, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Rybicki, B.A.; Johnson, C.C.; Uman, J.; Gorell, J.M. Parkinson’s disease mortality and the industrial use of heavy metals in michigan. Mov. Disord. 1993, 8, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposures to metals as risk factors for parkinson’s disease. Neurology 1997, 48, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of parkinson’s disease. Neurotoxicology 1999, 20, 239–247. [Google Scholar] [PubMed]
- Caudle, W.M. Occupational metal exposure and parkinsonism. Adv. Neurobiol. 2017, 18, 143–158. [Google Scholar]
- Bjorklund, G.; Stejskal, V.; Urbina, M.A.; Dadar, M.; Chirumbolo, S.; Mutter, J. Metals and parkinson’s disease: Mechanisms and biochemical processes. Curr. Med. Chem. 2018, 25, 2198–2214. [Google Scholar] [CrossRef]
- Letelier, M.E.; Faundez, M.; Jara-Sandoval, J.; Molina-Berrios, A.; Cortes-Troncoso, J.; Aracena-Parks, P.; Marin-Catalan, R. Mechanisms underlying the inhibition of the cytochrome p450 system by copper ions. J. Appl. Toxicol. 2009, 29, 695–702. [Google Scholar] [CrossRef]
- Scheuhammer, A.M.; Cherian, M.G. Effects of heavy metal cations, sulfhydryl reagents and other chemical agents on striatal d2 dopamine receptors. Biochem. Pharm. 1985, 34, 3405–3413. [Google Scholar] [CrossRef]
- Mosharov, E.V.; Borgkvist, A.; Sulzer, D. Presynaptic effects of levodopa and their possible role in dyskinesia. Mov. Disord. 2015, 30, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Bisaglia, M.; Filograna, R.; Beltramini, M.; Bubacco, L. Are dopamine derivatives implicated in the pathogenesis of parkinson’s disease? Ageing Res. Rev. 2014, 13, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, oxidative stress and protein-quinone modifications in parkinson’s and other neurodegenerative diseases. Angew Chem. Int. Ed. Engl. 2019, 58, 6512–6527. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; d’Ischia, M.; Misuraca, G.; Prota, G. Effect of metal ions on the rearrangement of dopachrome. Biochim. Biophys. Acta 1987, 925, 203–209. [Google Scholar] [CrossRef]
- Pham, A.N.; Waite, T.D. Cu(ii)-catalyzed oxidation of dopamine in aqueous solutions: Mechanism and kinetics. J. Inorg. Biochem. 2014, 137, 74–84. [Google Scholar] [CrossRef]
- Warren, P.J.; Earl, C.J.; Thompson, R.H. The distribution of copper in human brain. Brain 1960, 83, 709–717. [Google Scholar] [CrossRef]
- Davies, K.M.; Hare, D.J.; Cottam, V.; Chen, N.; Hilgers, L.; Halliday, G.; Mercer, J.F.; Double, K.L. Localization of copper and copper transporters in the human brain. Metallomics 2013, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Krebs, N.; Langkammer, C.; Goessler, W.; Ropele, S.; Fazekas, F.; Yen, K.; Scheurer, E. Assessment of trace elements in human brain using inductively coupled plasma mass spectrometry. J. Trace Elem. Med. Biol. 2014, 28, 1–7. [Google Scholar] [CrossRef]
- Dodani, S.C.; Domaille, D.W.; Nam, C.I.; Miller, E.W.; Finney, L.A.; Vogt, S.; Chang, C.J. Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and x-ray fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 5980–5985. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in parkinson’s disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Moghadam, A.; Charsouei, S.; Bell, B.; Jabalameli, M.R. Parkinson disease from mendelian forms to genetic susceptibility: New molecular insights into the neurodegeneration process. Cell Mol. Neurobiol. 2018, 38, 1153–1178. [Google Scholar] [CrossRef] [Green Version]
- Bisaglia, M.; Mammi, S.; Bubacco, L. Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J. 2009, 23, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.R.; Shin, H.J.; Lee, J.H.; Chang, C.S.; Kim, J. Copper(ii)-induced self-oligomerization of alpha-synuclein. Biochem. J. 1999, 340 Pt 3, 821–828. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular nk between parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [Green Version]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Interaction of alpha-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K. Cross talk between neurometals and amyloidogenic proteins at the synapse and the pathogenesis of neurodegenerative diseases. Metallomics 2017, 9, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.H.; Rospigliosi, C.; Eliezer, D. Nmr mapping of copper binding sites in alpha-synuclein. Biochim. Biophys. Acta 2006, 1764, 5–12. [Google Scholar] [CrossRef] [PubMed]
- McDowall, J.S.; Brown, D.R. Alpha-synuclein: Relating metals to structure, function and inhibition. Metallomics 2016, 8, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Structural characterization of copper(ii) binding to alpha-synuclein: Insights into the bioinorganic chemistry of parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef] [Green Version]
- Bharathi; Rao, K.S. Thermodynamics imprinting reveals differential binding of metals to alpha-synuclein: Relevance to parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 359, 115–120. [Google Scholar] [CrossRef]
- Binolfi, A.; Valiente-Gabioud, A.A.; Duran, R.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Exploring the structural details of cu(i) binding to alpha-synuclein by nmr spectroscopy. J. Am. Chem. Soc. 2011, 133, 194–196. [Google Scholar] [CrossRef]
- Bortolus, M.; Bisaglia, M.; Zoleo, A.; Fittipaldi, M.; Benfatto, M.; Bubacco, L.; Maniero, A.L. Structural characterization of a high affinity mononuclear site in the copper(ii)-alpha-synuclein complex. J. Am. Chem. Soc. 2010, 132, 18057–18066. [Google Scholar] [CrossRef] [PubMed]
- Dudzik, C.G.; Walter, E.D.; Millhauser, G.L. Coordination features and affinity of the cu(2)+ site in the alpha-synuclein protein of parkinson’s disease. Biochemistry 2011, 50, 1771–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valensin, D.; Dell’Acqua, S.; Kozlowski, H.; Casella, L. Coordination and redox properties of copper interaction with alpha-synuclein. J. Inorg. Biochem. 2016, 163, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Camponeschi, F.; Valensin, D.; Tessari, I.; Bubacco, L.; Dell’Acqua, S.; Casella, L.; Monzani, E.; Gaggelli, E.; Valensin, G. Copper(i)-alpha-synuclein interaction: Structural description of two independent and competing metal binding sites. Inorg. Chem. 2013, 52, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, A.S.; Ying, J.; Bax, A. Impact of n-terminal acetylation of alpha-synuclein on its random coil and lipid binding properties. Biochemistry 2012, 51, 5004–5013. [Google Scholar] [CrossRef]
- Dikiy, I.; Eliezer, D. N-terminal acetylation stabilizes n-terminal helicity in lipid- and micelle-bound alpha-synuclein and increases its affinity for physiological membranes. J. Biol. Chem. 2014, 289, 3652–3665. [Google Scholar] [CrossRef] [Green Version]
- Miotto, M.C.; Valiente-Gabioud, A.A.; Rossetti, G.; Zweckstetter, M.; Carloni, P.; Selenko, P.; Griesinger, C.; Binolfi, A.; Fernandez, C.O. Copper binding to the n-terminally acetylated, naturally occurring form of alpha-synuclein induces local helical folding. J. Am. Chem. Soc. 2015, 137, 6444–6447. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.J.; Paskins, A.R.; Dalton, C.F.; Smith, D.P. Copper binding and subsequent aggregation of alpha-synuclein are modulated by n-terminal acetylation and ablated by the h50q missense mutation. Biochemistry 2016, 55, 4737–4741. [Google Scholar] [CrossRef] [PubMed]
- Santner, A.; Uversky, V.N. Metalloproteomics and metal toxicology of alpha-synuclein. Metallomics 2010, 2, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Uversky, V.N. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics 2011, 3, 1163–1180. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Tessari, I.; Mammi, S.; Bubacco, L. Interaction between alpha-synuclein and metal ions, still looking for a role in the pathogenesis of parkinson’s disease. Neuromolecular Med. 2009, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Ilyechova, E.Y.; Miliukhina, I.V.; Orlov, I.A.; Muruzheva, Z.M.; Puchkova, L.V.; Karpenko, M.N. A low blood copper concentration is a co-morbidity burden factor in parkinson’s disease development. Neurosci. Res. 2018, 135, 54–62. [Google Scholar] [CrossRef]
- Kim, M.J.; Oh, S.B.; Kim, J.; Kim, K.; Ryu, H.S.; Kim, M.S.; Ayton, S.; Bush, A.I.; Lee, J.Y.; Chung, S.J. Association of metals with the risk and clinical characteristics of parkinson’s disease. Parkinsonism Relat. Disord. 2018, 55, 117–121. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef]
- Uitti, R.J.; Rajput, A.H.; Rozdilsky, B.; Bickis, M.; Wollin, T.; Yuen, W.K. Regional metal concentrations in parkinson’s disease, other chronic neurological diseases, and control brains. Can. J. Neurol. Sci. 1989, 16, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, D.A.; LeWitt, P.A.; Juneau, P.L.; Sima, A.A.; Nguyen, H.U.; DeMaggio, A.J.; Brickman, C.M.; Brewer, G.J.; Dick, R.D.; Troyer, M.D.; et al. Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res. 1996, 738, 265–274. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for parkinson disease. Ann. Neurol. 2013, 73, 554–559. [Google Scholar] [CrossRef]
- Davies, K.M.; Bohic, S.; Carmona, A.; Ortega, R.; Cottam, V.; Hare, D.J.; Finberg, J.P.; Reyes, S.; Halliday, G.M.; Mercer, J.F.; et al. Copper pathology in vulnerable brain regions in parkinson’s disease. Neurobiol. Aging 2014, 35, 858–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, Z.L.; Klomp, L.W.; Gitlin, J.D. Aceruloplasminemia: An inherited neurodegenerative disease with impairment of iron homeostasis. Am. J. Clin. Nutr. 1998, 67, 972S–977S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.P.; David, S. Ceruloplasmin regulates iron levels in the cns and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Pin, S.; Gathinji, M.; Fuchs, R.; Harris, Z.L. Aceruloplasminemia: An inherited neurodegenerative disease with impairment of iron homeostasis. Ann. NY Acad. Sci. 2004, 1012, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.F.; Prohaska, J.R.; Knutson, M.D. Metabolic crossroads of iron and copper. Nutr. Rev. 2010, 68, 133–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J.G.; et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in parkinson’s disease brain. Acta Neuropathol. 2017, 134, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Murayama, S.; Shimizu, J.; Ohya, Y.; Kwak, S.; Asayama, K.; Kanazawa, I. Cu/zn superoxide dismutase-like immunoreactivity is present in lewy bodies from parkinson disease: A light and electron microscopic immunocytochemical study. Acta Neuropathol. 1995, 89, 471–474. [Google Scholar] [CrossRef]
- Roudeau, S.; Chevreux, S.; Carmona, A.; Ortega, R. Reduced net charge and heterogeneity of pi isoforms in familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase. Electrophoresis 2015, 36, 2482–2488. [Google Scholar] [CrossRef]
- Filograna, R.; Beltramini, M.; Bubacco, L.; Bisaglia, M. Anti-oxidants in parkinson’s disease therapy: A critical point of view. Curr. Neuropharmacol. 2016, 14, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Moreau, C.; Duce, J.A.; Rascol, O.; Devedjian, J.C.; Berg, D.; Dexter, D.; Cabantchik, Z.I.; Bush, A.I.; Devos, D. Iron as a therapeutic target for parkinson’s disease. Mov. Disord. 2018, 33, 568–574. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Grunblatt, E.; Mandel, S. The copper chelator, d-penicillamine, does not attenuate mptp induced dopamine depletion in mice. J. Neural. Transm. (Vienna) 2007, 114, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Rios, C.; Alvarez-Vega, R.; Rojas, P. Depletion of copper and manganese in brain after mptp treatment of mice. Pharm. Toxicol. 1995, 76, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, D.S.; Pinheiro, A.B.; Castineiras-Filho, S.L.; da Silva, A.S.; Miotto, M.C.; De Falco, A.; de, P.R.T.; Maisonette, S.; da Cunha, A.L.; Hauser-Davis, R.A.; et al. A moderate metal-binding hydrazone meets the criteria for a bioinorganic approach towards parkinson’s disease: Therapeutic potential, blood-brain barrier crossing evaluation and preliminary toxicological studies. J. Inorg. Biochem. 2017, 170, 160–168. [Google Scholar] [CrossRef]
- Roberts, B.R.; Lim, N.K.; McAllum, E.J.; Donnelly, P.S.; Hare, D.J.; Doble, P.A.; Turner, B.J.; Price, K.A.; Lim, S.C.; Paterson, B.M.; et al. Oral treatment with cu(ii)(atsm) increases mutant sod1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 8021–8031. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.R.; Trias, E.; Beilby, P.R.; Lopez, N.I.; Labut, E.M.; Bradford, C.S.; Roberts, B.R.; McAllum, E.J.; Crouch, P.J.; Rhoads, T.W.; et al. Copper delivery to the cns by cuatsm effectively treats motor neuron disease in sod(g93a) mice co-expressing the copper-chaperone-for-sod. Neurobiol. Dis. 2016, 89, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hilton, J.B.; Mercer, S.W.; Lim, N.K.; Faux, N.G.; Buncic, G.; Beckman, J.S.; Roberts, B.R.; Donnelly, P.S.; White, A.R.; Crouch, P.J. Cu(ii)(atsm) improves the neurological phenotype and survival of sod1(g93a) mice and selectively increases enzymatically active sod1 in the spinal cord. Sci. Rep. 2017, 7, 42292. [Google Scholar] [CrossRef] [Green Version]
- Vavere, A.L.; Lewis, J.S. Cu-atsm: A radiopharmaceutical for the pet imaging of hypoxia. Dalton Trans. 2007, 4893–4902. [Google Scholar] [CrossRef]
- Yoshii, Y.; Yoneda, M.; Ikawa, M.; Furukawa, T.; Kiyono, Y.; Mori, T.; Yoshii, H.; Oyama, N.; Okazawa, H.; Saga, T.; et al. Radiolabeled cu-atsm as a novel indicator of overreduced intracellular state due to mitochondrial dysfunction: Studies with mitochondrial DNA-less rho0 cells and cybrids carrying melas mitochondrial DNA mutation. Nucl. Med. Biol. 2012, 39, 177–185. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Kudo, T.; Kuriyama, M.; Fujibayashi, Y.; Yoneda, M. Evaluation of striatal oxidative stress in patients with parkinson’s disease using [62cu]atsm pet. Nucl. Med. Biol. 2011, 38, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S.; et al. The hypoxia imaging agent cuii(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of parkinson’s disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisaglia, M.; Bubacco, L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules 2020, 10, 195. https://doi.org/10.3390/biom10020195
Bisaglia M, Bubacco L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules. 2020; 10(2):195. https://doi.org/10.3390/biom10020195
Chicago/Turabian StyleBisaglia, Marco, and Luigi Bubacco. 2020. "Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant?" Biomolecules 10, no. 2: 195. https://doi.org/10.3390/biom10020195
APA StyleBisaglia, M., & Bubacco, L. (2020). Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules, 10(2), 195. https://doi.org/10.3390/biom10020195