Follow the Mutations: Toward Class-Specific, Small-Molecule Reactivation of p53

Abstract
:1. Introduction
2. Crosstalk Between Mutational Classes
3. Rescuing Stability Class Mutants: Cavity Binders
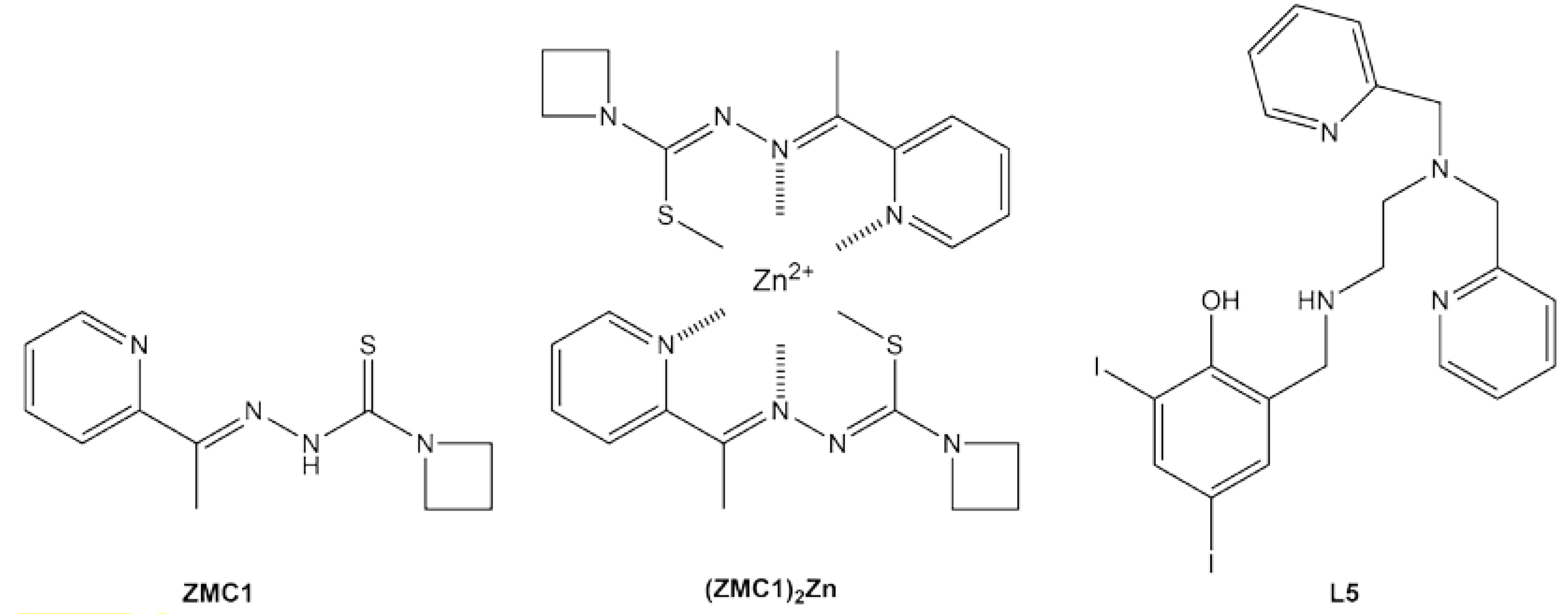
4. Rescuing Zinc-Binding Class Mutants: Zinc Metallochaperones
5. Rescuing DNA Contact Mutants
6. Rescuing p53 Aggregation
7. Alkylating Agents
8. Challenges and Outlook
Funding
Conflicts of Interest
References
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumor types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.-K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Shahbandi, A.; Jackson, J.G. Analysis across multiple tumor types provides no evidence that mutant p53 exerts dominant negative activity. NPJ Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Moll, U.M. Depleting stabilized GOF mutant p53 proteins by inhibiting molecular folding chaperones: A new promise in cancer therapy. Cell Death Differ. 2017, 24, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Tahaney, W.M.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecularly targeted therapies for p53-mutant cancers. Cell. Mol. Life Sci. 2017, 74, 4171–4187. [Google Scholar] [CrossRef]
- Raj, N.; Attardi, L.D. The Transactivation Domains of the p53 Protein. Cold Spring Harb Perspect Med. 2017, 7. [Google Scholar] [CrossRef]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, J.S.; Loh, S.N. Structure, function, and aggregation of the zinc-free form of the p53 DNA binding domain. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Blanden, A.R.; Narayanan, S.; Jayakumar, L.; Lubin, D.; Augeri, D.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget 2014, 5, 8879–8892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.-B.; DeDecker, B.S.; Freund, S.M.; Proctor, M.R.; Bycroft, M.; Fersht, A.R. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA 1999, 96, 8438–8442. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.S.; Loh, S.N. Folding and misfolding mechanisms of the p53 DNA binding domain at physiological temperature. Protein Sci. 2006, 15, 2457–2465. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.S.; Loh, S.N. Zn2+-dependent misfolding of the p53 DNA binding domain. Biochemistry 2007, 46, 2630–2639. [Google Scholar] [CrossRef]
- Butler, J.S.; Loh, S.N. Kinetic partitioning during folding of the p53 DNA binding domain. J. Mol. Biol. 2005, 350, 906–918. [Google Scholar] [CrossRef]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Ghosh, D.; Ranganathan, S.; Anoop, A.; Santosh Kuma, P.; Jha, N.N.; Padinhateeri, R.; Maji, S.K. Investigating the Intrinsic Aggregation Potential of Evolutionarily Conserved Segments in p53. Biochemistry 2014, 53, 5995–6010. [Google Scholar] [CrossRef]
- Ishimaru, D.; Andrade, L.R.; Teixeira, L.S.P.; Quesado, P.A.; Maiolino, L.M.; Lopez, P.M.; Cordeiro, Y.; Costa, L.T.; Heckl, W.M.; Weissmüller, G.; et al. Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry 2003, 42, 9022–9027. [Google Scholar] [CrossRef]
- Wilcken, R.; Wang, G.; Boeckler, F.M.; Fersht, A.R. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. USA 2012, 109, 13584–13589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bom, A.P.D.A.; Rangel, L.P.; Costa, D.C.F.; de Oliveira, G.A.P.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.I.; Cepeda, A.O.T.; Stumbo, A.C.; et al. Mutant p53 Aggregates into Prion-like Amyloid Oligomers and Fibrils IMPLICATIONS FOR CANCER. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smet, F.D.; Rubio, M.S.; Hompes, D.; Naus, E.; Baets, G.D.; Langenberg, T.; Hipp, M.S.; Houben, B.; Claes, F.; Charbonneau, S.; et al. Nuclear inclusion bodies of mutant and wild-type p53 in cancer: A hallmark of p53 inactivation and proteostasis remodelling by p53 aggregation. J. Pathol. 2017, 242, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic. Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Baud, M.G.J.; Wilcken, R.; Boeckler, F.M.; Fersht, A.R.; Joerger, A.C.; Spencer, J. Harnessing Fluorine–Sulfur Contacts and Multipolar Interactions for the Design of p53 Mutant Y220C Rescue Drugs. ACS Chem. Biol. 2016, 11, 2265–2274. [Google Scholar] [CrossRef] [Green Version]
- Wilcken, R.; Liu, X.; Zimmermann, M.O.; Rutherford, T.J.; Fersht, A.R.; Joerger, A.C.; Boeckler, F.M. Halogen-Enriched Fragment Libraries as Leads for Drug Rescue of Mutant p53. J. Am. Chem. Soc. 2012, 134, 6810–6818. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.A.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med.Chem. 2019, 11, 2491–2504. [Google Scholar] [CrossRef] [Green Version]
- Baud, M.G.J.; Bauer, M.R.; Verduci, L.; Dingler, F.A.; Patel, K.J.; Horil Roy, D.; Joerger, A.C.; Fersht, A.R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018, 152, 101–114. [Google Scholar] [CrossRef]
- Wassman, C.D.; Baronio, R.; Demir, O.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef]
- Bromley, D.; Bauer, M.R.; Fersht, A.R.; Daggett, V. An in silico algorithm for identifying stabilizing pockets in proteins: Test case, the Y220C mutant of the p53 tumor suppressor protein. Protein Eng. Des. Sel. 2016, 29, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joerger, A.C.; Bauer, M.R.; Wilcken, R.; Baud, M.G.J.; Harbrecht, H.; Exner, T.E.; Boeckler, F.M.; Spencer, J.; Fersht, A.R. Exploiting Transient Protein States for the Design of Small-Molecule Stabilizers of Mutant p53. Structure 2015, 23, 2246–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic. Acids Res. 2019, 47, 1637–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, S.N. The missing zinc: p53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Na, B.; Yu, X.; Withers, T.; Gilleran, J.; Yao, M.; Foo, T.K.; Chen, C.; Moore, D.; Lin, Y.; Kimball, S.D.; et al. Therapeutic targeting of BRCA1 and TP53 mutant breast cancer through mutant p53 reactivation. NPJ Breast Cancer 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Kogan, S.; Chen, Y.; Tsang, A.T.; Withers, T.; Lin, H.; Gilleran, J.; Buckley, B.; Moore, D.; Bertino, J.; et al. Zinc Metallochaperones Reactivate Mutant p53 Using an ON/OFF Switch Mechanism: A New Paradigm in Cancer Therapeutics. Clin. Cancer Res. 2018, 24, 4505–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Blanden, A.; Tsang, A.T.; Zaman, S.; Liu, Y.; Gilleran, J.; Bencivenga, A.F.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53. Mol. Pharmacol. 2017, 91, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, A.; Patel, A.A.; Tang, L.; Liu, Z.; Wang, L.; Silver, N.L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Reznik, E.; Stokes, M.E.; Krishnamoorthy, L.; Bos, P.H.; Song, Y.; Quartararo, C.E.; Pagano, N.C.; Carpizo, D.R.; deCarvalho, A.C.; et al. Copper-Binding Small Molecule Induces Oxidative Stress and Cell-Cycle Arrest in Glioblastoma-Patient-Derived Cells. Cell Chem. Biol. 2018, 25, 585–594.e7. [Google Scholar] [CrossRef]
- Zaman, S.; Yu, X.; Bencivenga, A.F.; Blanden, A.R.; Liu, Y.; Withers, T.; Na, B.; Blayney, A.J.; Gilleran, J.; Boothman, D.A.; et al. Combinatorial therapy of Zinc metallochaperones with mutant p53 reactivation and diminished copper binding. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef]
- Miller, J.J.; Orvain, C.; Jozi, S.; Clarke, R.M.; Smith, J.R.; Blanchet, A.; Gaiddon, C.; Warren, J.J.; Storr, T. Multifunctional Compounds for Activation of the p53-Y220C Mutant in Cancer. Chemistry A 2018, 24, 17734–17742. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Ang, H.C.; Veprintsev, D.B.; Blair, C.M.; Fersht, A.R. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 2005, 280, 16030–16037. [Google Scholar] [CrossRef] [Green Version]
- Baroni, T.E.; Wang, T.; Qian, H.; Dearth, L.R.; Truong, L.N.; Zeng, J.; Denes, A.E.; Chen, S.W.; Brachmann, R.K. A global suppressor motif for p53 cancer mutants. Proc. Natl. Acad. Sci. USA 2004, 101, 4930–4935. [Google Scholar] [CrossRef] [Green Version]
- Rasquinha, J.A.; Bej, A.; Dutta, S.; Mukherjee, S. Intrinsic Differences in Backbone Dynamics between Wild Type and DNA-Contact Mutants of the p53 DNA Binding Domain Revealed by Nuclear Magnetic Resonance Spectroscopy. Biochemistry 2017, 56, 4962–4971. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.S.; Ramos, H.; Gomes, S.; Loureiro, J.B.; Soares, J.; Barcherini, V.; Monti, P.; Fronza, G.; Oliveira, C.; Domingues, L.; et al. SLMP53-1 interacts with wild-type and mutant p53 DNA-binding domain and reactivates multiple hotspot mutations. Biochim. Biophys. Acta 2020, 1864, 129440. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Raimundo, L.; Pereira, N.A.L.; Monteiro, Â.; Gomes, S.; Bessa, C.; Pereira, C.; Queiroz, G.; Bisio, A.; Fernandes, J.; et al. Reactivation of wild-type and mutant p53 by tryptophanolderived oxazoloisoindolinone SLMP53-1, a novel anticancer small-molecule. Oncotarget 2016, 7, 4326–4343. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.F.; de Oliveira, A.P.G. Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer. Acc. Chem. Res. 2018, 51, 181–190. [Google Scholar] [CrossRef]
- Navalkar, A.; Ghosh, S.; Pandey, S.; Paul, A.; Datta, D.; Maji, S.K. Prion-like p53 Amyloids in Cancer. Biochemistry 2019. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, L.; Chang, Y.; Li, Y.; Butler, W.; Jin, E.; Wang, A.; Tao, Y.; Chen, X.; Liang, C.; et al. Therapeutic potential of ReACp53 targeting mutant p53 protein in CRPC. Prostate Cancer Prostatic Dis. 2019. [Google Scholar] [CrossRef]
- Ishimaru, D.; Ano Bom, A.P.; Lima, L.M.; Quesado, P.A.; Oyama, M.F.; de Moura Gallo, C.V.; Cordeiro, Y.; Silva, J.L. Cognate DNA stabilizes the tumor suppressor p53 and prevents misfolding and aggregation. Biochemistry 2009, 48, 6126–6135. [Google Scholar] [CrossRef]
- Silva, J.L.; Vieira, T.C.R.G.; Gomes, M.P.B.; Bom, A.P.A.; Lima, L.M.T.R.; Freitas, M.S.; Ishimaru, D.; Cordeiro, Y.; Foguel, D. Ligand Binding and Hydration in Protein Misfolding: Insights from Studies of Prion and p53 Tumor Suppressor Proteins. Acc. Chem. Res. 2010, 43, 271–279. [Google Scholar] [CrossRef]
- Kovachev, P.S.; Banerjee, D.; Rangel, L.P.; Eriksson, J.; Pedrote, M.M.; Martins-Dinis, M.M.D.C.; Edwards, K.; Cordeiro, Y.; Silva, J.L.; Sanyal, S. Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain. J. Biol. Chem. 2017, 292, 9345–9357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Chen, J.; Keshamouni, V.G.; Kanapathipillai, M. Polyarginine and its analogues inhibit p53 mutant aggregation and cancer cell proliferation in vitro. Biochem. Biophy. Res. Commun. 2017, 489, 130–134. [Google Scholar] [CrossRef] [PubMed]
- da Costa, D.C.F.; Campos, N.P.C.; Santos, R.A.; Guedes-da-Silva, F.H.; Martins-Dinis, M.M.D.C.; Zanphorlin, L.; Ramos, C.; Rangel, L.P.; Silva, J.L. Resveratrol prevents p53 aggregation in vitro and in breast cancer cells. Oncotarget 2018, 9, 29112–29122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rasitnejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system x C-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Zhang, M.Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.; Arner, E.S.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Zhang, Q.; Bergman, J.; Wiman, K.G.; Bykov, V.J.N. Role of Thiol Reactivity for Targeting Mutant p53. Cell Chem. Biol. 2018, 25, 1219–1230.e3. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omar, S.I.; Tuszynski, J. The molecular mechanism of action of methylene quinuclidinone and its effects on the structure of p53 mutants. Oncotarget 2018, 9, 37137–37156. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.R.; Joerger, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, E.; Parker, T.M.; Bauer, M.R.; Dhiman, A.; Pelham, C.J.; Nagane, M.; Kuppusamy, M.L.; Holmes, M.; Holmes, T.R.; Shaik, K.; et al. The curcumin analog HO-3867 selectively kills cancer cells by converting mutant p53 protein to transcriptionally active wildtype p53. J. Biol. Chem. 2018, 293, 4262–4276. [Google Scholar] [CrossRef] [Green Version]
- Rangel, L.P.; Ferretti, G.D.S.; Costa, C.L.; Andrade, S.M.M.V.; Carvalho, R.S.; Costa, D.C.F.; Silva, J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [Green Version]
- Synnott, N.C.; Bauer, M.R.; Madden, S.; Murray, A.; Klinger, R.; O’Donovan, N.; O’Connor, D.; Gallagher, W.M.; Crown, J.; Fersht, A.R.; et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018, 414, 99–106. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-C.; He, L.; Khoshaba, R.; Lu, F.-G.; Cai, C.; Zhou, F.-L.; Liao, D.-F.; Cao, D. Curcumin Nicotinate Selectively Induces Cancer Cell Apoptosis and Cycle Arrest through a P53-Mediated Mechanism. Molecules 2019, 24, 4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | WT | Mutants | DNA Contact | Zinc Binding | Stability | Ref. |
|---|---|---|---|---|---|---|
| 1 | R248 | Gln, Trp | Yes | No | No | [20,22] |
| 2 | R273 | His, Cys | Yes | No | No | [20] |
| 3 | R175 | His | No | Yes | No | [23] |
| 4 | G245 | Ser, Asp, Cys, Val | No | Yes | Yes | [20,23] |
| 5 | R249 | Ser | Yes | Yes | [20,24] | |
| 6 | Y220 | Cys | No | Yes | [20] | |
| 7 | C176 | Phe, Tyr | No | Yes | [23],1 | |
| 8 | H179 | Arg, Tyr | No | Yes | 1 | |
| 9 | V157 | Phe | No | Yes | [20] | |
| 10 | M237 | Ile | No | Yes | [20] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, S.N. Follow the Mutations: Toward Class-Specific, Small-Molecule Reactivation of p53. Biomolecules 2020, 10, 303. https://doi.org/10.3390/biom10020303
Loh SN. Follow the Mutations: Toward Class-Specific, Small-Molecule Reactivation of p53. Biomolecules. 2020; 10(2):303. https://doi.org/10.3390/biom10020303
Chicago/Turabian StyleLoh, Stewart N. 2020. "Follow the Mutations: Toward Class-Specific, Small-Molecule Reactivation of p53" Biomolecules 10, no. 2: 303. https://doi.org/10.3390/biom10020303
APA StyleLoh, S. N. (2020). Follow the Mutations: Toward Class-Specific, Small-Molecule Reactivation of p53. Biomolecules, 10(2), 303. https://doi.org/10.3390/biom10020303