Activins as Dual Specificity TGF-β Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
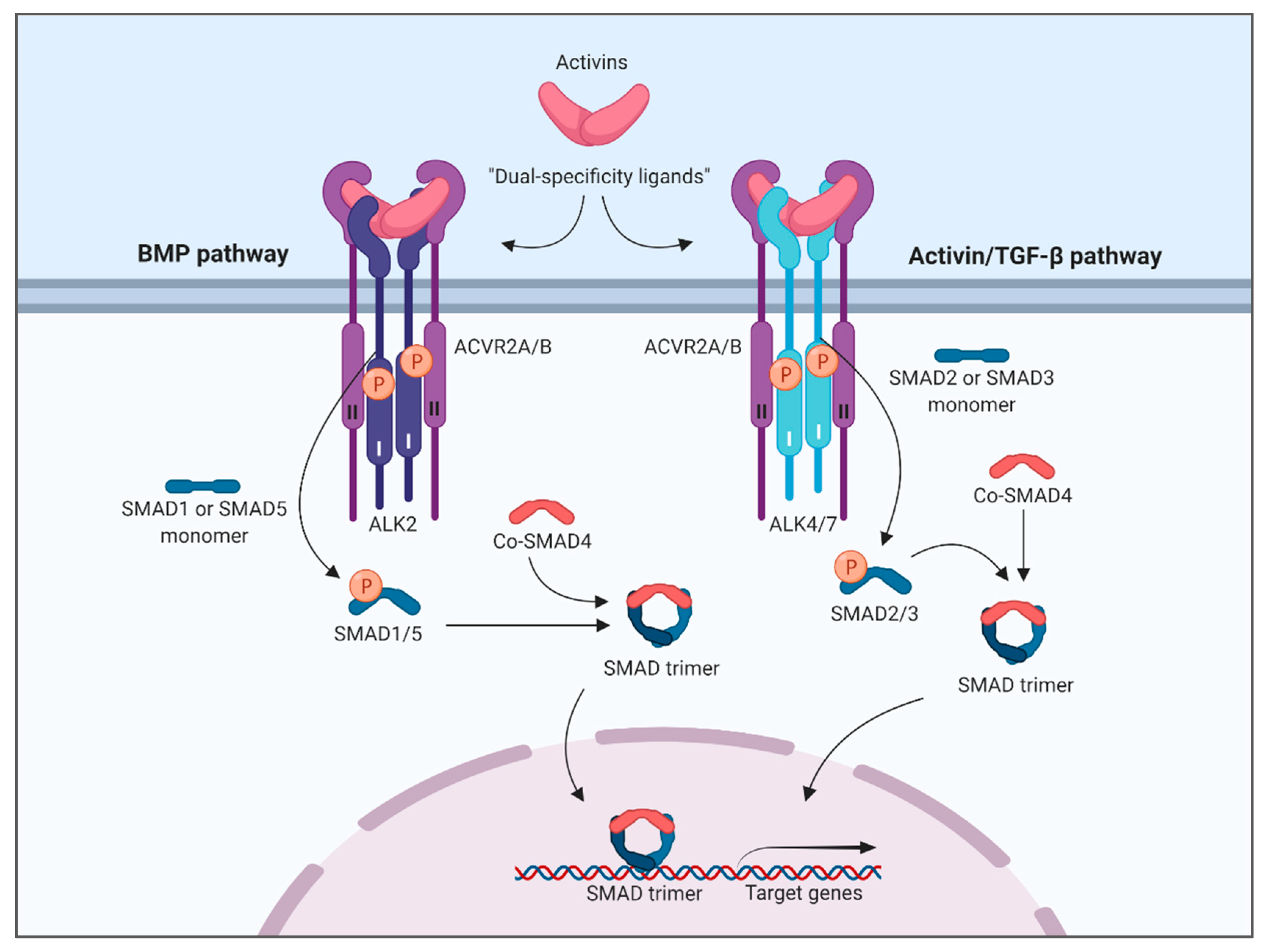
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Western Blotting
2.3. Cell Viability
2.4. Transfection of INA-6 Cells
2.5. Transfection of HepG2 Cells
2.6. Comparative RT-PCR
2.7. Statistical Analysis
3. Results
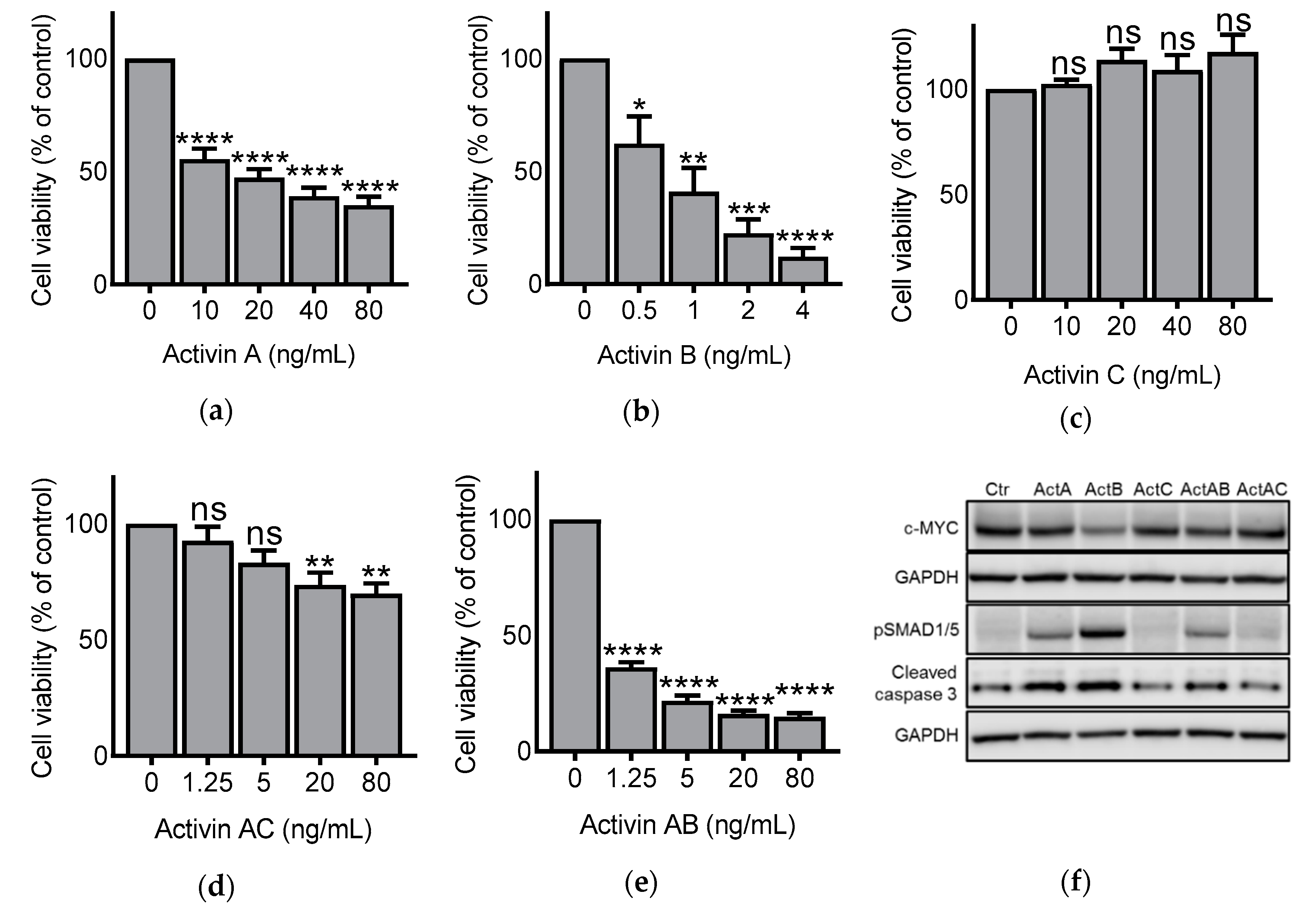
3.1. Activin Dimers Have Dose-Dependent Effects on IH-1 Cell Viability
3.2. Activins Activated SMAD1/5 and SMAD2 with Different Dynamics
3.3. Effect of Small Molecule Inhibitors on Activin-Induced SMAD Phosphorylation
3.4. Effect of the BMP Type 1 Receptor Inhibitor, K02288, on Activin-Induced SMAD Activity in HepG2 Cells
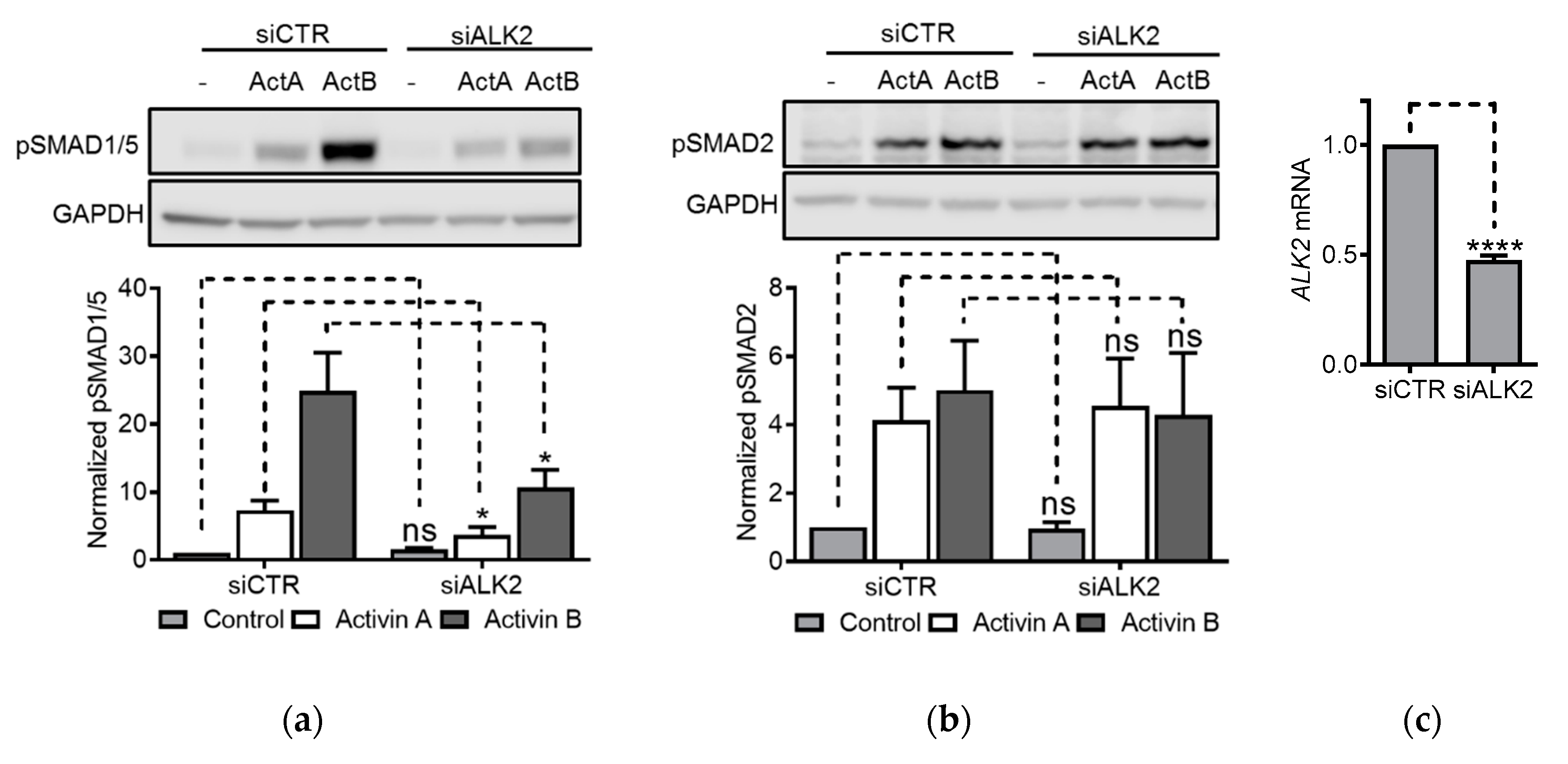
3.5. ALK2 Knockdown Blunted Activin-Induced SMAD1/5 Activity
3.6. Antagonism of Activins by Follistatin and Cerberus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010, 147, 35–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Yamashita, H.; Ichijo, H.; Franzen, P.; Laiho, M.; Miyazono, K.; Heldin, C.H. Characterization of type I receptors for transforming growth factor-beta and activin. Science 1994, 264, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Nakatani, M.; Yamakawa, N.; Hashimoto, O.; Hasegawa, Y.; Sugino, H. Activin isoforms signal through type I receptor serine/threonine kinase ALK7. Mol. Cell. Endocrinol. 2004, 220, 59–65. [Google Scholar] [CrossRef]
- Attisano, L.; Carcamo, J.; Ventura, F.; Weis, F.M.; Massague, J.; Wrana, J.L. Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell 1993, 75, 671–680. [Google Scholar] [CrossRef]
- Ebner, R.; Chen, R.H.; Lawler, S.; Zioncheck, T.; Derynck, R. Determination of type I receptor specificity by the type II receptors for TGF-beta or activin. Science 1993, 262, 900–902. [Google Scholar] [CrossRef]
- Piek, E.; Afrakhte, M.; Sampath, K.; van Zoelen, E.J.; Heldin, C.H.; ten Dijke, P. Functional antagonism between activin and osteogenic protein-1 in human embryonal carcinoma cells. J. Cell Physiol. 1999, 180, 141–149. [Google Scholar] [CrossRef]
- Olsen, O.E.; Wader, K.F.; Hella, H.; Mylin, A.K.; Turesson, I.; Nesthus, I.; Waage, A.; Sundan, A.; Holien, T. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun. Signal. 2015, 13, 27. [Google Scholar] [CrossRef] [Green Version]
- Aykul, S.; Martinez-Hackert, E. Transforming Growth Factor-beta Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J. Biol. Chem. 2016, 291, 10792–10804. [Google Scholar] [CrossRef] [Green Version]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besson-Fournier, C.; Latour, C.; Kautz, L.; Bertrand, J.; Ganz, T.; Roth, M.P.; Coppin, H. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 2012, 120, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canali, S.; Core, A.B.; Zumbrennen-Bullough, K.B.; Merkulova, M.; Wang, C.Y.; Schneyer, A.L.; Pietrangelo, A.; Babitt, J.L. Activin B Induces Noncanonical SMAD1/5/8 Signaling via BMP Type I Receptors in Hepatocytes: Evidence for a Role in Hepcidin Induction by Inflammation in Male Mice. Endocrinology 2016, 157, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Haupt, J.; Xu, M.; Shore, E.M. Variable signaling activity by FOP ACVR1 mutations. Bone 2017, 109, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.E.; Sankar, M.; Elsaadi, S.; Hella, H.; Buene, G.; Darvekar, S.R.; Misund, K.; Katagiri, T.; Knaus, P.; Holien, T. BMPR2 inhibits activin- and BMP-signaling via wild type ALK2. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Holien, T.; Vatsveen, T.K.; Hella, H.; Rampa, C.; Brede, G.; Groseth, L.A.; Rekvig, M.; Borset, M.; Standal, T.; Waage, A.; et al. Bone morphogenetic proteins induce apoptosis in multiple myeloma cells by Smad-dependent repression of MYC. Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund UK 2012, 26, 1073–1080. [Google Scholar] [CrossRef]
- Holien, T.; Sundan, A. The role of bone morphogenetic proteins in myeloma cell survival. Cytokine Growth Factor Rev. 2014, 25, 343–350. [Google Scholar] [CrossRef]
- Holien, T.; Vatsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-MYC in multiple myeloma. Blood 2012, 120, 2450–2453. [Google Scholar] [CrossRef] [Green Version]
- Hjertner, O.; Hjorth-Hansen, H.; Borset, M.; Seidel, C.; Waage, A.; Sundan, A. Bone morphogenetic protein-4 inhibits proliferation and induces apoptosis of multiple myeloma cells. Blood 2001, 97, 516–522. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.; Guenther, A.; Bakker, F.; Schmalzing, M.; Bernand, S.; Baum, W.; Duerr, B.; Hocke, G.M.; Steininger, H.; Gebhart, E.; et al. Gp130 and ras mediated signaling in human plasma cell line INA-6: A cytokine-regulated tumor model for plasmacytoma. Hematol. J. Off. J. Eur. Haematol. Assoc. 2001, 2, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.E.; Morris-Triggs, S.A.; Ruotolo, B.T.; Robinson, C.V.; Ohnuma, S.; Hyvonen, M. Structural basis for the inhibition of activin signalling by follistatin. Embo J. 2006, 25, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aykul, S.; Ni, W.; Mutatu, W.; Martinez-Hackert, E. Human Cerberus prevents nodal-receptor binding, inhibits nodal signaling, and suppresses nodal-mediated phenotypes. PLoS ONE 2015, 10, e0114954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.H.; Sreenu, D.; Krishnaiah, M.; Subrahmanyam, V.B.; Rao, K.S.; Nagendra Mohan, A.V.; Park, C.Y.; Son, J.Y.; Son, D.H.; Park, H.J.; et al. Synthesis and biological evaluation of 1-substituted-3(5)-(6-methylpyridin-2-yl)-4-(quinoxalin-6-yl)pyrazoles as transforming growth factor-beta type 1 receptor kinase inhibitors. Eur. J. Med. Chem. 2011, 46, 3917–3925. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.E.; Wader, K.F.; Misund, K.; Vatsveen, T.K.; Ro, T.B.; Mylin, A.K.; Turesson, I.; Stordal, B.F.; Moen, S.H.; Standal, T.; et al. Bone morphogenetic protein-9 suppresses growth of myeloma cells by signaling through ALK2 but is inhibited by endoglin. Blood Cancer J. 2014, 4, e196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerli, U.M.; Holt, R.U.; Holien, T.; Vaatsveen, T.K.; Zhan, F.; Egeberg, K.W.; Barlogie, B.; Waage, A.; Aarset, H.; Dai, H.Y.; et al. Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood 2008, 111, 806–815. [Google Scholar] [CrossRef]
- Baughn, L.B.; Di Liberto, M.; Niesvizky, R.; Cho, H.J.; Jayabalan, D.; Lane, J.; Liu, F.; Chen-Kiang, S. CDK2 phosphorylation of Smad2 disrupts TGF-beta transcriptional regulation in resistant primary bone marrow myeloma cells. J. Immunol. 2009, 182, 1810–1817. [Google Scholar] [CrossRef] [Green Version]
- Sanvitale, C.E.; Kerr, G.; Chaikuad, A.; Ramel, M.C.; Mohedas, A.H.; Reichert, S.; Wang, Y.; Triffitt, J.T.; Cuny, G.D.; Yu, P.B.; et al. A new class of small molecule inhibitor of BMP signaling. PLoS ONE 2013, 8, e62721. [Google Scholar] [CrossRef] [Green Version]
- Engers, D.W.; Frist, A.Y.; Lindsley, C.W.; Hong, C.C.; Hopkins, C.R. Synthesis and structure-activity relationships of a novel and selective bone morphogenetic protein receptor (BMP) inhibitor derived from the pyrazolo[1.5-a]pyrimidine scaffold of dorsomorphin: The discovery of ML347 as an ALK2 versus ALK3 selective MLPCN probe. Bioorg. Med. Chem. Lett. 2013, 23, 3248–3252. [Google Scholar] [CrossRef] [Green Version]
- Cuny, G.D.; Yu, P.B.; Laha, J.K.; Xing, X.; Liu, J.F.; Lai, C.S.; Deng, D.Y.; Sachidanandan, C.; Bloch, K.D.; Peterson, R.T. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4388–4392. [Google Scholar] [CrossRef] [Green Version]
- Inman, G.J.; Nicolas, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef]
- Gellibert, F.; Woolven, J.; Fouchet, M.H.; Mathews, N.; Goodland, H.; Lovegrove, V.; Laroze, A.; Nguyen, V.L.; Sautet, S.; Wang, R.; et al. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-beta type I receptor inhibitors. J. Med. Chem. 2004, 47, 4494–4506. [Google Scholar] [CrossRef] [PubMed]
- Ro, T.B.; Holt, R.U.; Brenne, A.T.; Hjorth-Hansen, H.; Waage, A.; Hjertner, O.; Sundan, A.; Borset, M. Bone morphogenetic protein-5, -6 and -7 inhibit growth and induce apoptosis in human myeloma cells. Oncogene 2004, 23, 3024–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Takio, K.; Eto, Y.; Shibai, H.; Titani, K.; Sugino, H. Activin-binding protein from rat ovary is follistatin. Science 1990, 247, 836–838. [Google Scholar] [CrossRef] [PubMed]
- Schneyer, A.; Schoen, A.; Quigg, A.; Sidis, Y. Differential binding and neutralization of activins A and B by follistatin and follistatin like-3 (FSTL-3/FSRP/FLRG). Endocrinology 2003, 144, 1671–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenster, U.; Harrison, C.A.; Donaldson, C.; Vale, W.; Fischer, W.H. An activin-A/C chimera exhibits activin and myostatin antagonistic properties. J. Biol. Chem. 2005, 280, 36626–36632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccolo, S.; Agius, E.; Leyns, L.; Bhattacharyya, S.; Grunz, H.; Bouwmeester, T.; De Robertis, E.M. The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature 1999, 397, 707–710. [Google Scholar] [CrossRef]
- Aykul, S.; Martinez-Hackert, E. New Ligand Binding Function of Human Cerberus and Role of Proteolytic Processing in Regulating Ligand-Receptor Interactions and Antagonist Activity. J. Mol. Biol. 2016, 428, 590–602. [Google Scholar] [CrossRef]
- Wakefield, L.M.; Hill, C.S. Beyond TGFbeta: Roles of other TGFbeta superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L.; Montalvo, E.; Massague, J. Activation of signalling by the activin receptor complex. Mol. Cell. Biol. 1996, 16, 1066–1073. [Google Scholar] [CrossRef] [Green Version]
- Mellor, S.L.; Cranfield, M.; Ries, R.; Pedersen, J.; Cancilla, B.; de Kretser, D.; Groome, N.P.; Mason, A.J.; Risbridger, G.P. Localization of activin beta(A)-, beta(B)-, and beta(C)-subunits in humanprostate and evidence for formation of new activin heterodimers of beta(C)-subunit. J. Clin. Endocrinol. Metab. 2000, 85, 4851–4858. [Google Scholar] [CrossRef] [Green Version]
- Mellor, S.L.; Ball, E.M.; O’Connor, A.E.; Ethier, J.F.; Cranfield, M.; Schmitt, J.F.; Phillips, D.J.; Groome, N.P.; Risbridger, G.P. Activin betaC-subunit heterodimers provide a new mechanism of regulating activin levels in the prostate. Endocrinology 2003, 144, 4410–4419. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T.; Sebald, W.; Dreyer, M.K. Crystal structure of the BMP-2-BRIA ectodomain complex. Nat. Struct. Biol. 2000, 7, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Goebel, E.J.; Corpina, R.A.; Hinck, C.S.; Czepnik, M.; Castonguay, R.; Grenha, R.; Boisvert, A.; Miklossy, G.; Fullerton, P.T.; Matzuk, M.M.; et al. Structural characterization of an activin class ternary receptor complex reveals a third paradigm for receptor specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 15505–15513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idone, V.; Corpina, R.; Goebel, E.; Cunanan, C.; Dimitriou, A.; Kim, H.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. The finger 2 tip loop of Activin A is required for the formation of its non-signaling complex with ACVR1 and type II Bone Morphogenetic Protein receptors. bioRxiv 2019. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-beta Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Daly, A.C.; Randall, R.A.; Hill, C.S. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell. Biol. 2008, 28, 6889–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, A.; Vizan, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Muller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-beta uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. eLife 2018, 7, e31756. [Google Scholar] [CrossRef]
- Miller, D.S.J.; Schmierer, B.; Hill, C.S. TGF-beta family ligands exhibit distinct signalling dynamics that are driven by receptor localisation. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Vizan, P.; Miller, D.S.; Gori, I.; Das, D.; Schmierer, B.; Hill, C.S. Controlling long-term signaling: Receptor dynamics determine attenuation and refractory behavior of the TGF-beta pathway. Sci. Signal. 2013, 6, ra106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, D.L.; Sapkota, G.P. Phosphatases in SMAD regulation. Febs Lett. 2012, 586, 1897–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traeger, L.; Gallitz, I.; Sekhri, R.; Baumer, N.; Kuhlmann, T.; Kemming, C.; Holtkamp, M.; Muller, J.C.; Karst, U.; Canonne-Hergaux, F.; et al. ALK3 undergoes ligand-independent homodimerization and BMP-induced heterodimerization with ALK2. Free Radic. Biol. Med. 2018, 129, 127–137. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olsen, O.E.; Hella, H.; Elsaadi, S.; Jacobi, C.; Martinez-Hackert, E.; Holien, T. Activins as Dual Specificity TGF-β Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors. Biomolecules 2020, 10, 519. https://doi.org/10.3390/biom10040519
Olsen OE, Hella H, Elsaadi S, Jacobi C, Martinez-Hackert E, Holien T. Activins as Dual Specificity TGF-β Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors. Biomolecules. 2020; 10(4):519. https://doi.org/10.3390/biom10040519
Chicago/Turabian StyleOlsen, Oddrun Elise, Hanne Hella, Samah Elsaadi, Carsten Jacobi, Erik Martinez-Hackert, and Toril Holien. 2020. "Activins as Dual Specificity TGF-β Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors" Biomolecules 10, no. 4: 519. https://doi.org/10.3390/biom10040519
APA StyleOlsen, O. E., Hella, H., Elsaadi, S., Jacobi, C., Martinez-Hackert, E., & Holien, T. (2020). Activins as Dual Specificity TGF-β Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors. Biomolecules, 10(4), 519. https://doi.org/10.3390/biom10040519