Calculation of the Geometries and Infrared Spectra of the Stacked Cofactor Flavin Adenine Dinucleotide (FAD) as the Prerequisite for Studies of Light-Triggered Proton and Electron Transfer


Abstract
:1. Introduction
2. Materials and Methods
3. Results
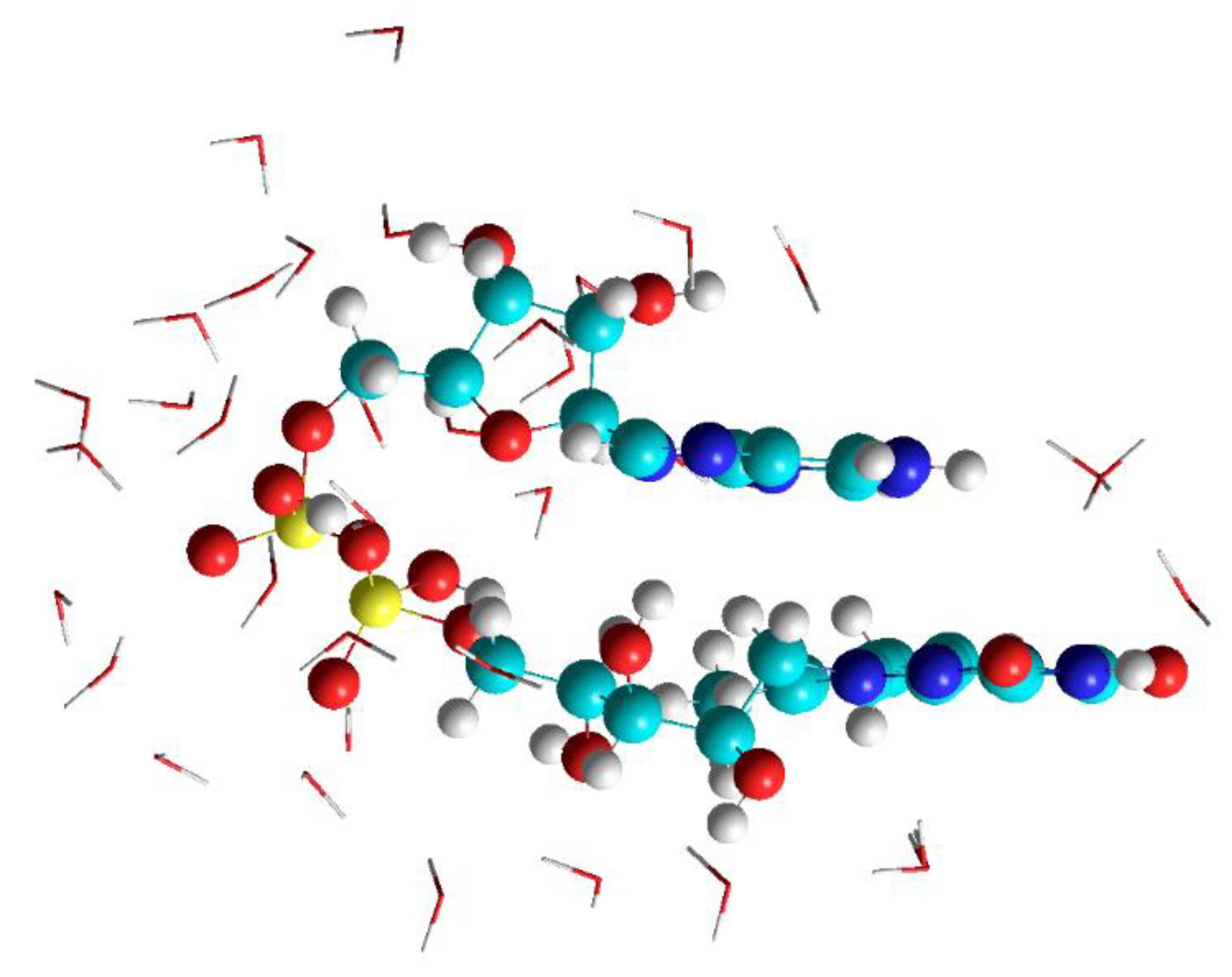
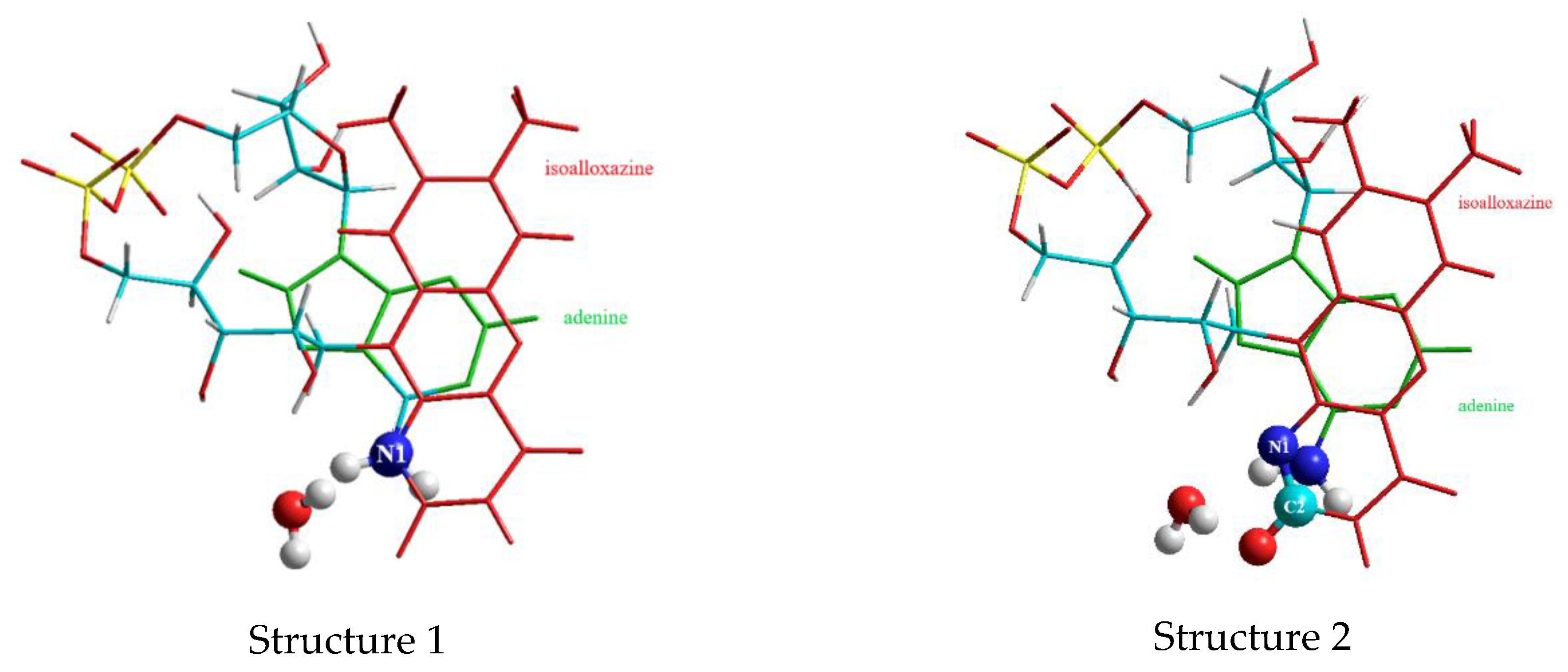
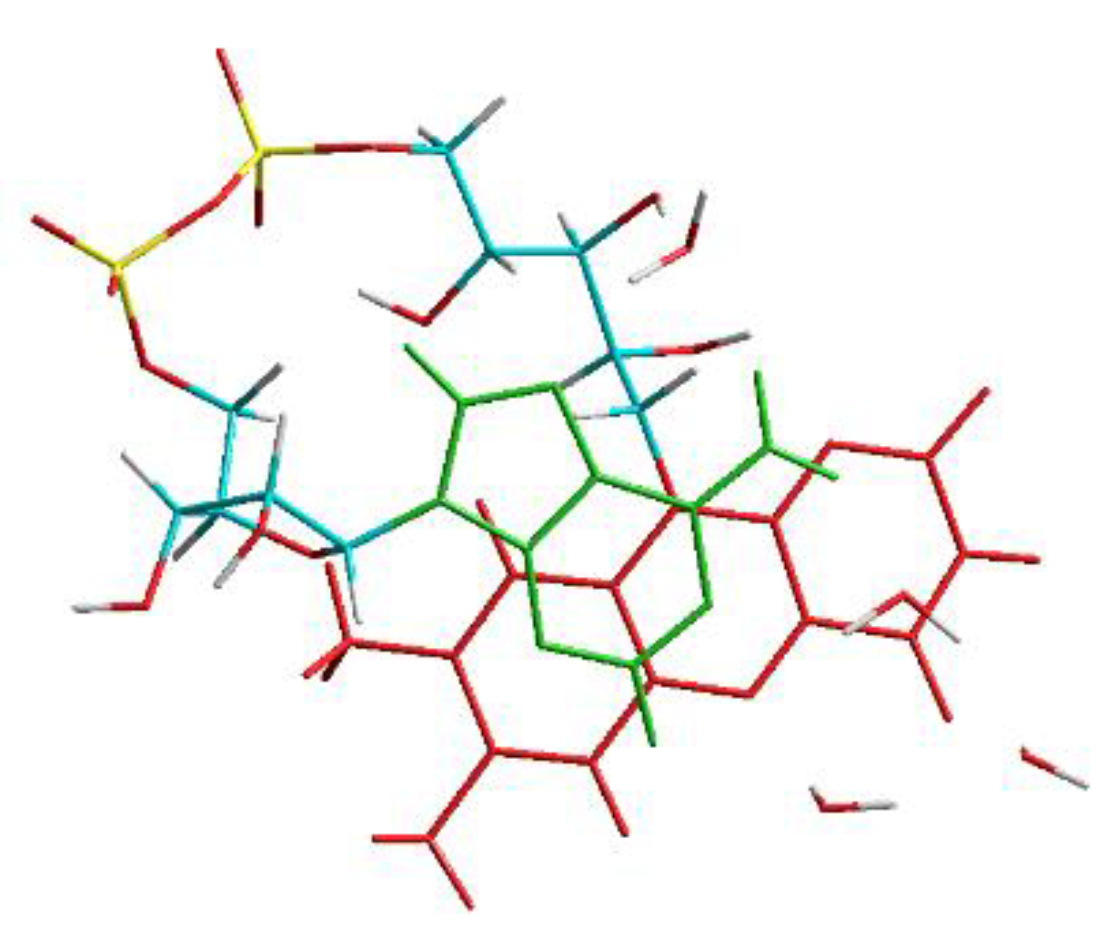
3.1. MD Calculations to Obtain Starting Geometries of FAD in the Closed Geometry
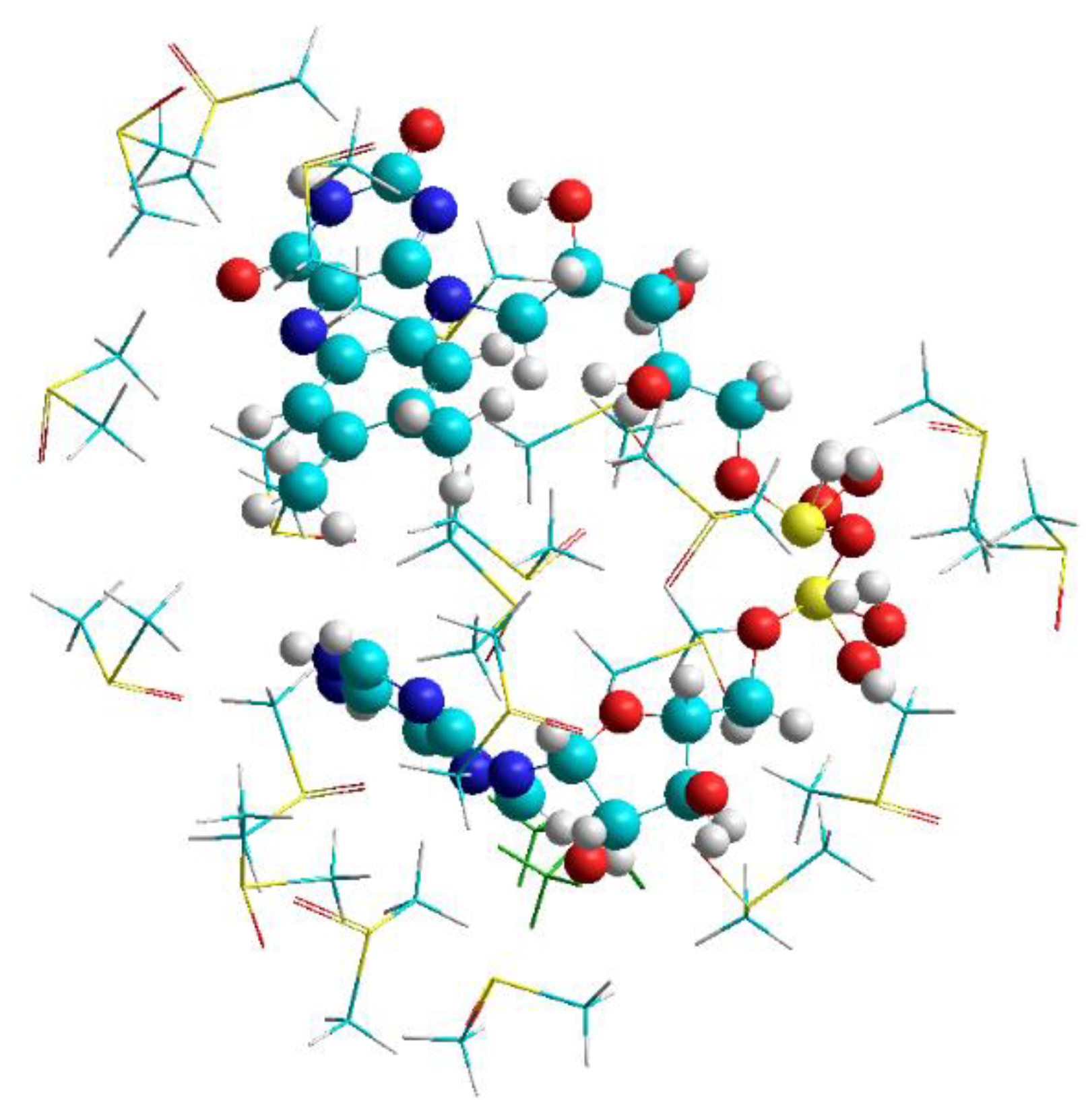
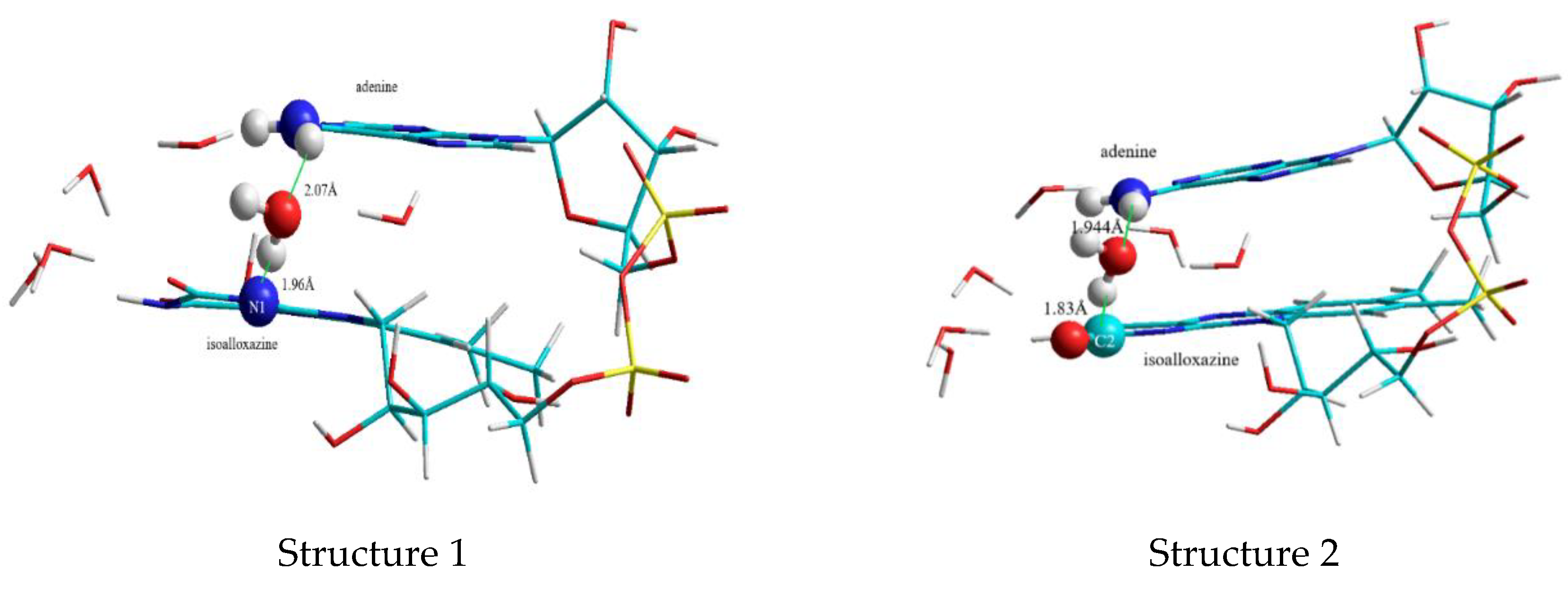
3.2. DFT Studies on the Closed Complex FAD-Water and the Role of the Water Molecules in the FAD-Water Complex
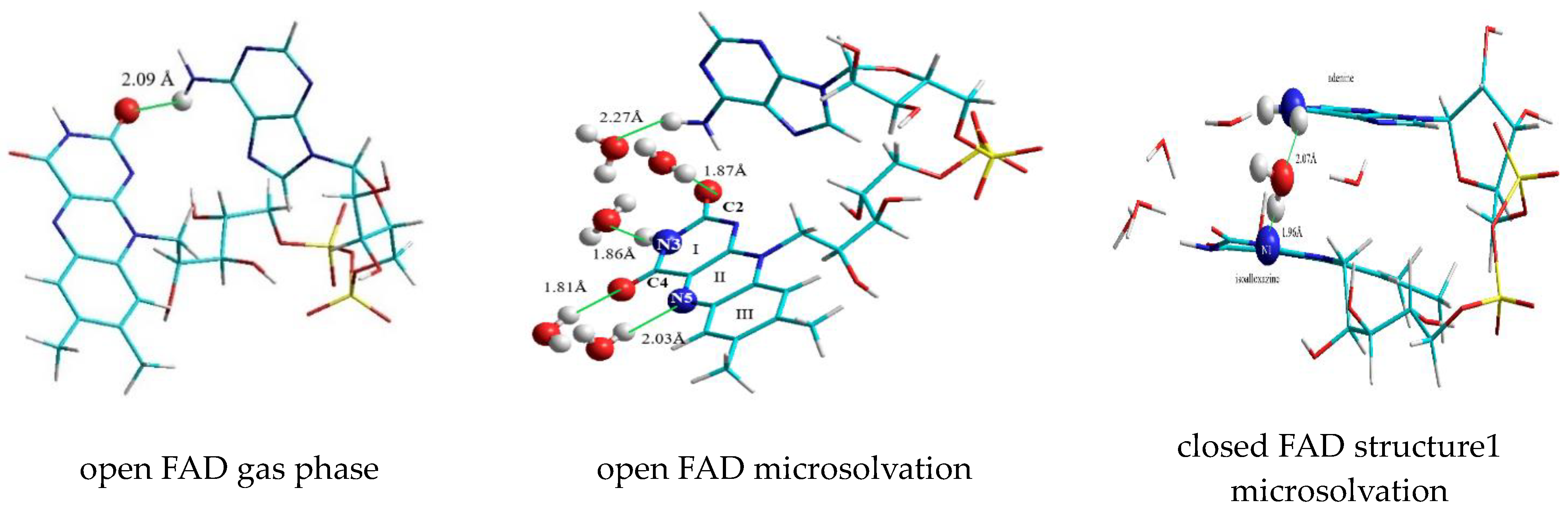
3.2.1. The First “Water Clip”
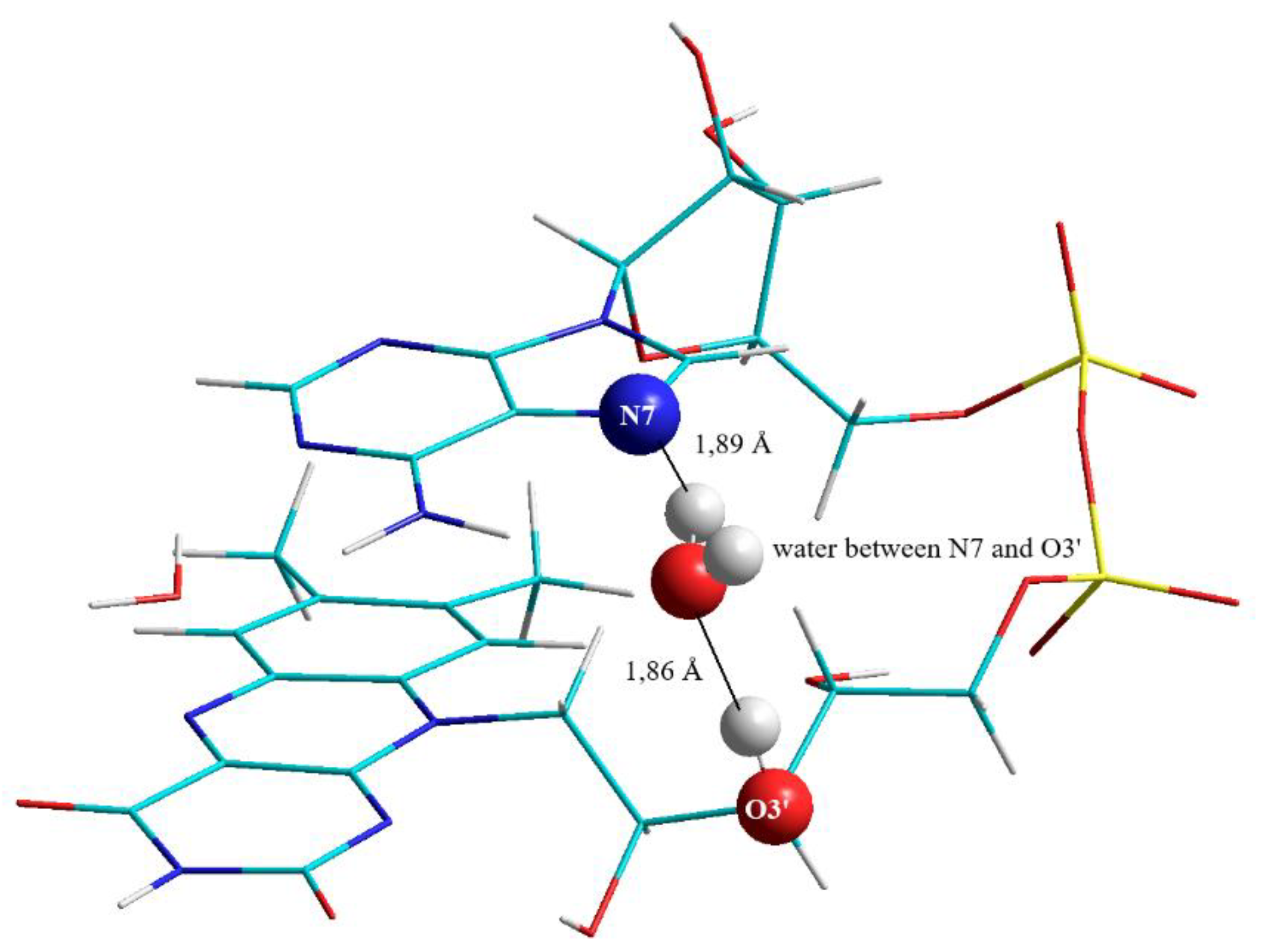
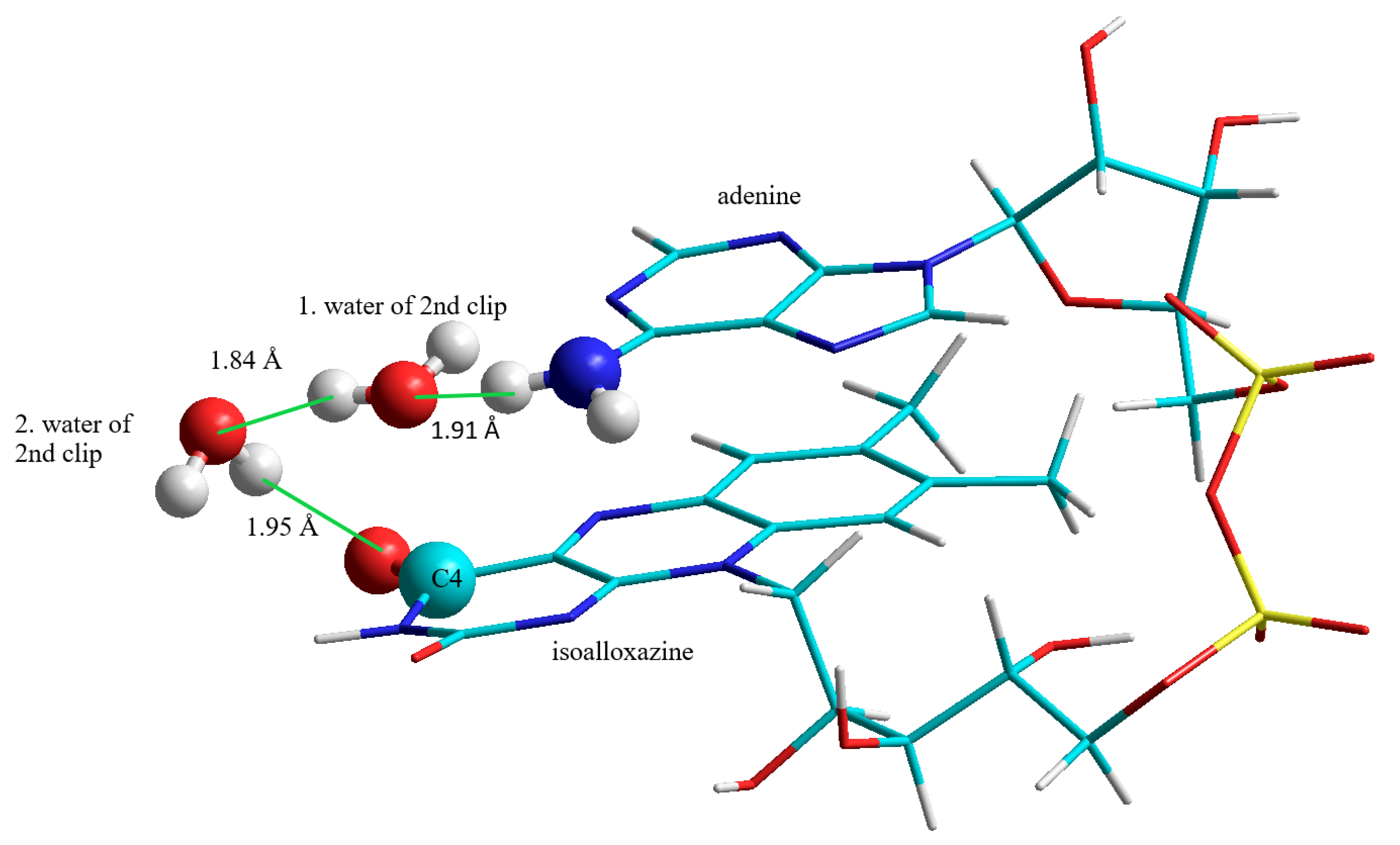
3.2.2. The Second “Water Clip”
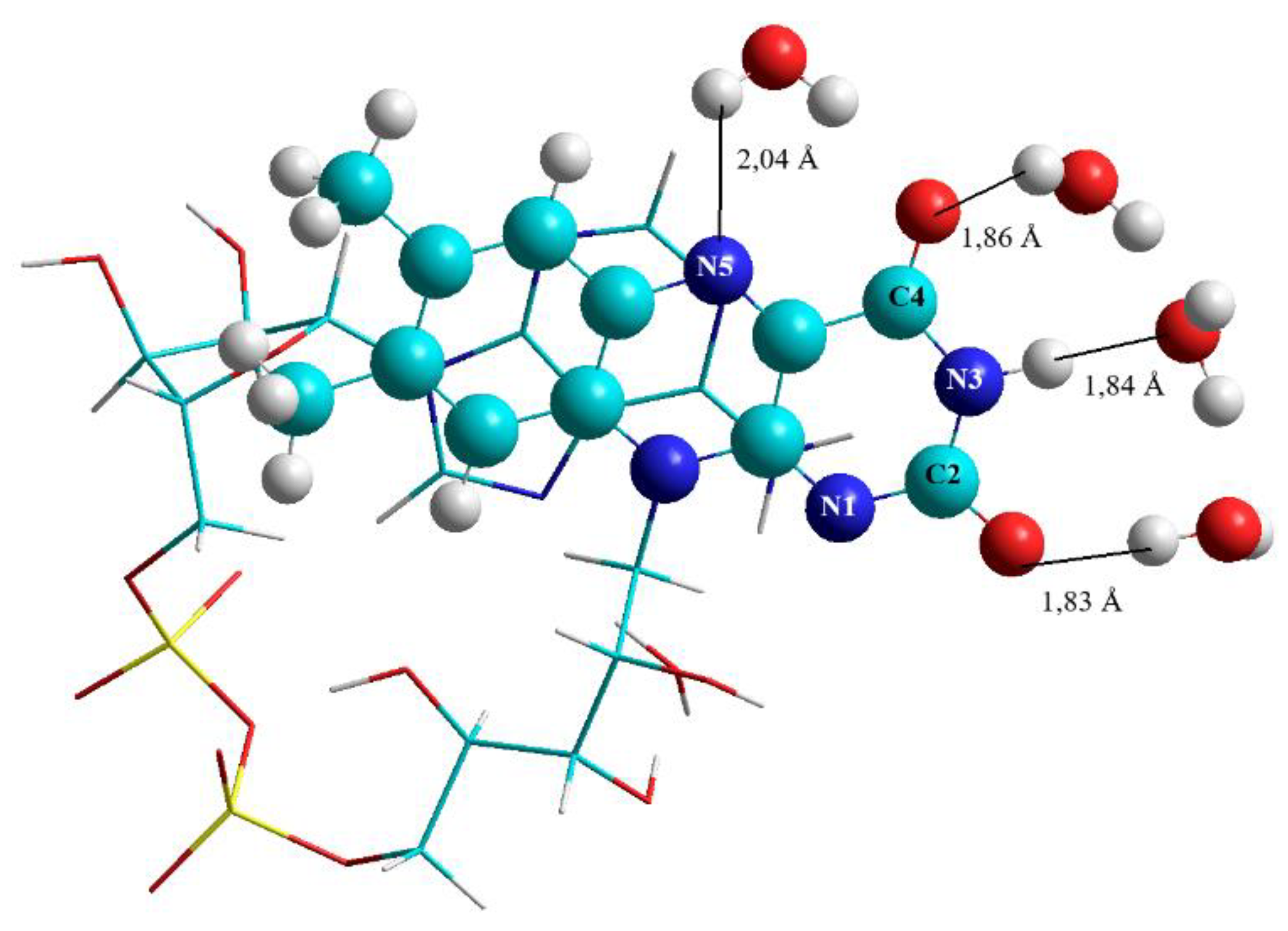
3.2.3. Water Molecules Surrounding Isoalloxazine at the C2, N3, C4, and N5 Positions
3.2.4. The Third “Water Clip”
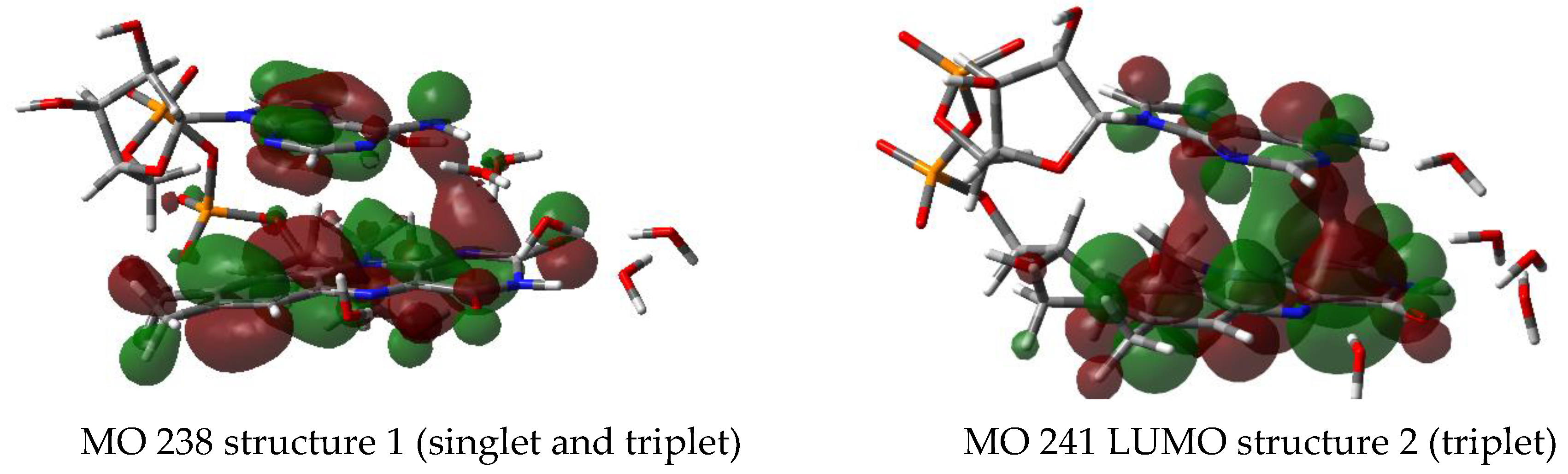
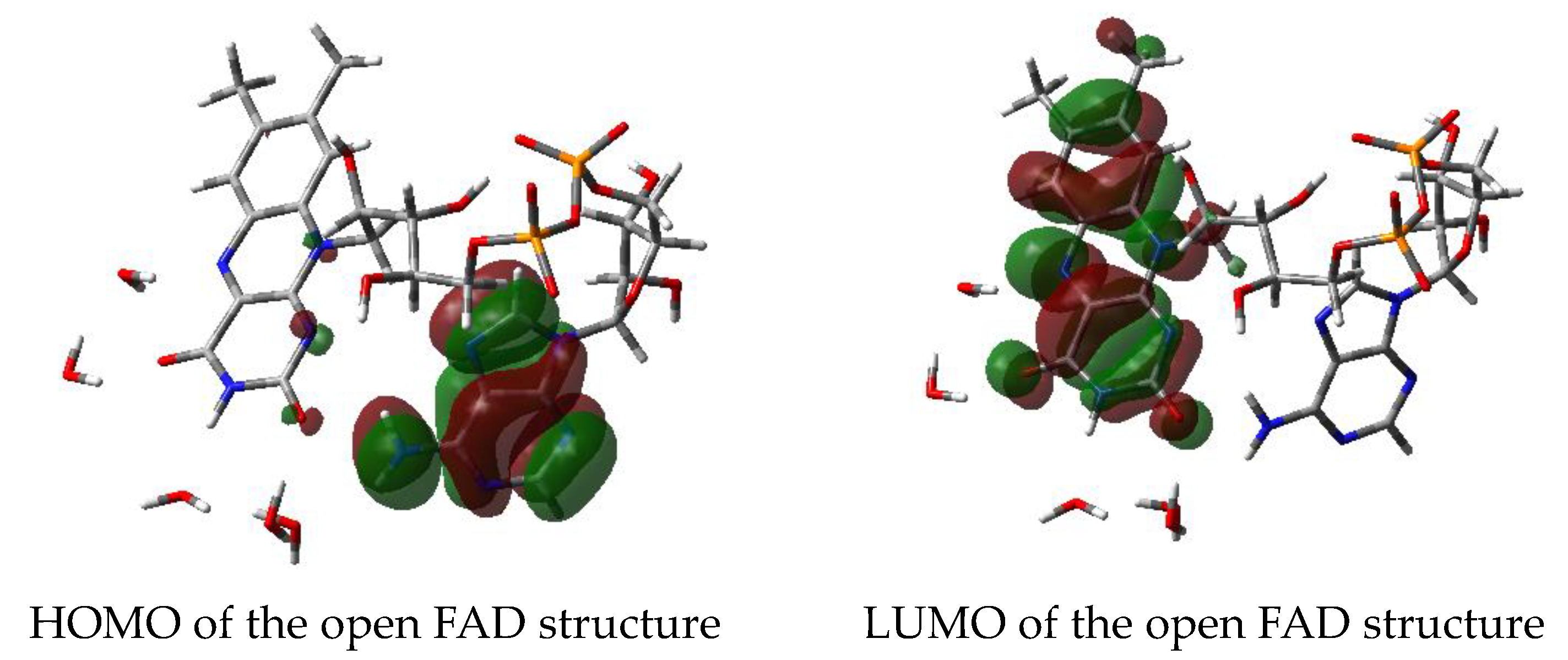
3.2.5. Molecular Orbitals—HOMO/LUMO
3.2.6. Supramolecular Orbitals
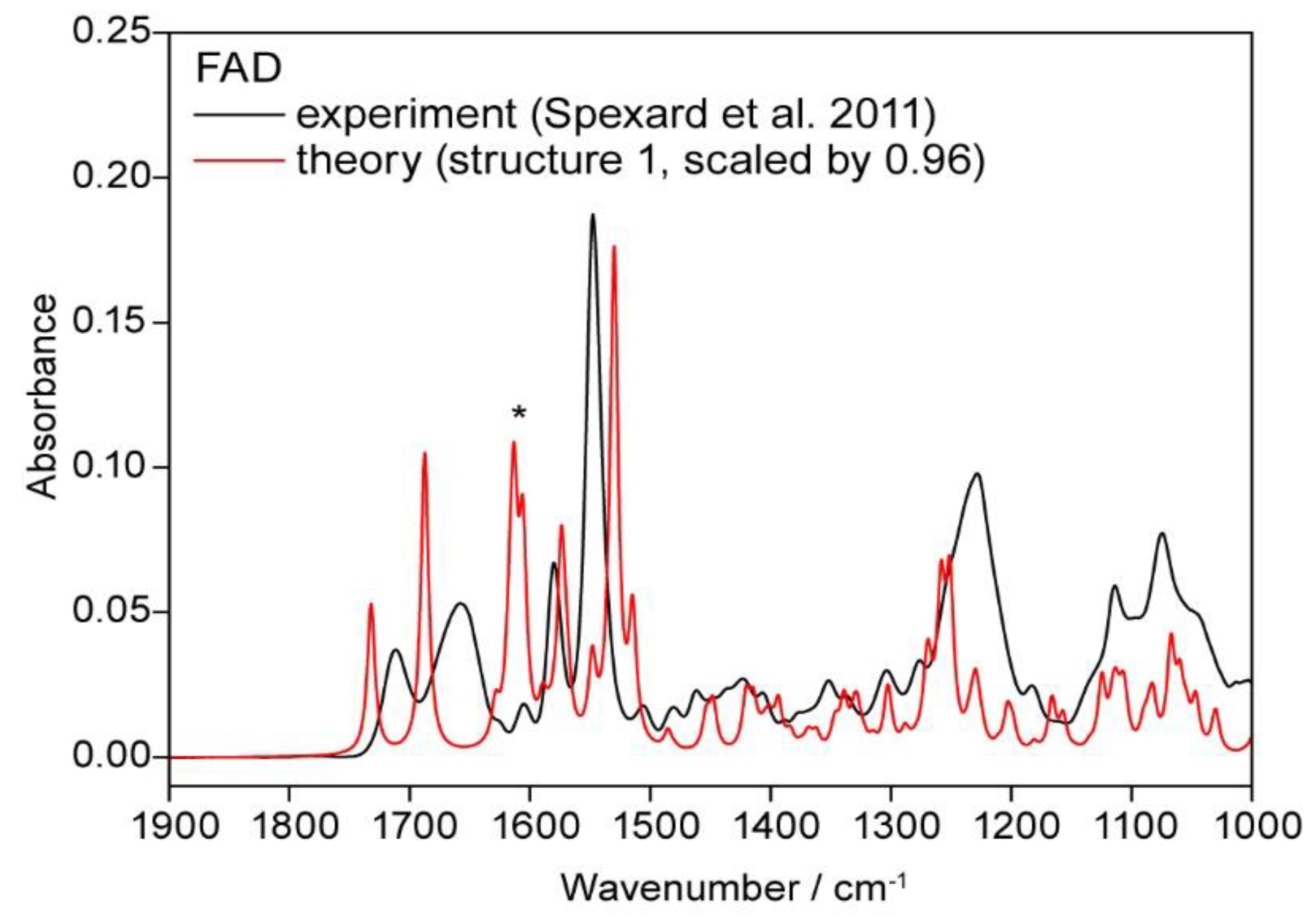
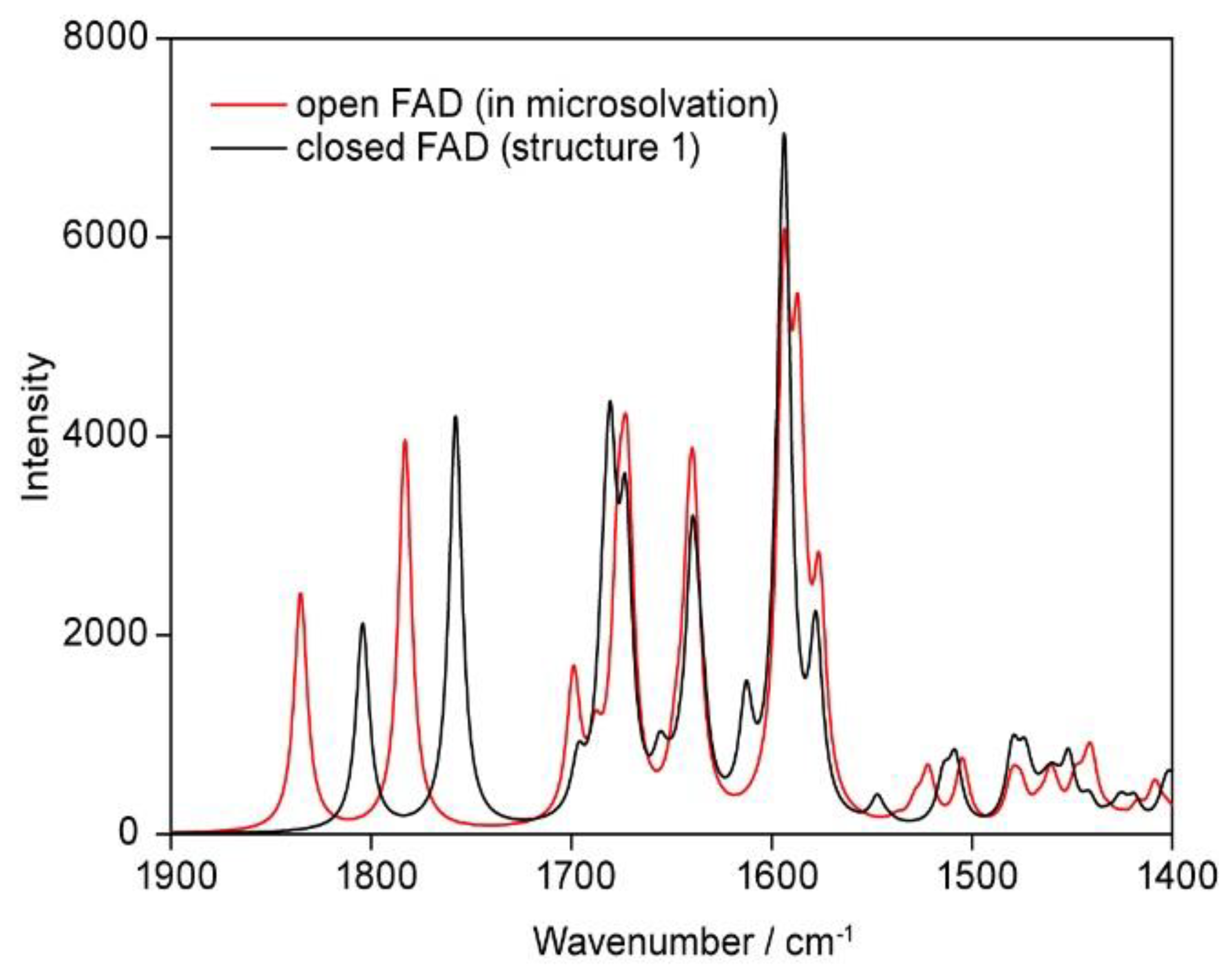
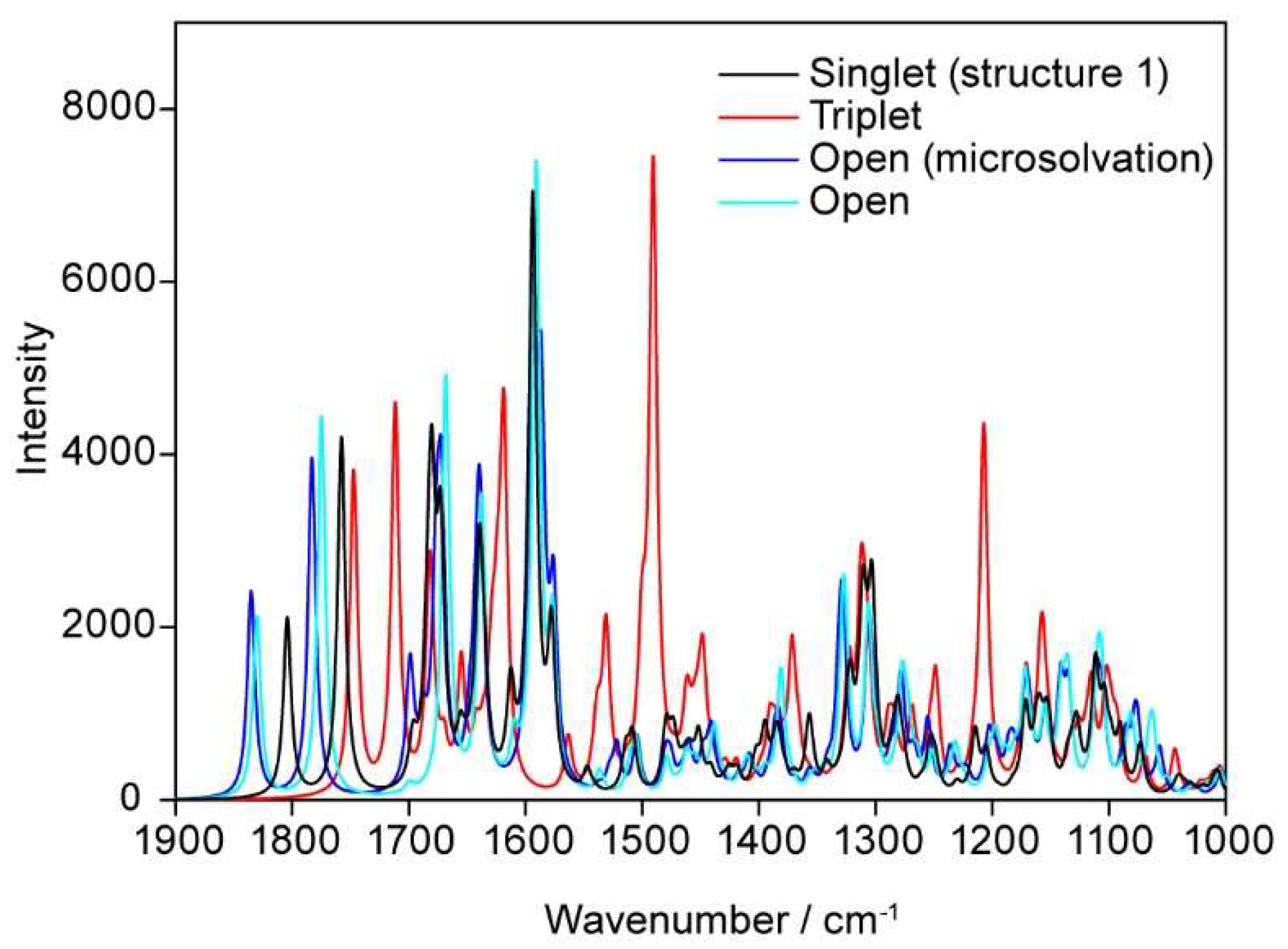
3.2.7. Vibrational Spectra
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Losi, A.; Gärtner, W. Solving Blue Light Riddles: New Lessons from Flavin-binding LOV Photoreceptors. Photochem. Photobiol. 2017, 93, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Chaves, I.; Pokorny, R.; Byrdin, M.; Hoang, N.; Ritz, T.; Brettel, K.; Essen, L.O.; van der Horst, G.T.; Batschauer, A.; Ahmad, M. The cryptochromes: Blue light photoreceptors in plants and animals. Annu. Rev. Plant. Biol. 2011, 62, 335–364. [Google Scholar] [CrossRef] [PubMed]
- Losi, A.; Gärtner, W. The evolution of flavin-binding photoreceptors: An ancient chromophore serving trendy blue-light sensors. Annu. Rev. Plant. Biol. 2012, 63, 49–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegear, R.J.; Casselman, A.; Waddell, S.; Reppert, S.M. Cryptochrome mediates light-dependent magnetosensitivity in Drosophila. Nature 2008, 454, 1014–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, A.; Einwich, A.; Sjulstok, E.; Feederle, R.; Bolte, P.; Koch, K.; Solov’yov, I.A.; Mouritsen, H. Double-Cone Localization and Seasonal Expression Pattern Suggest a Role in Magnetoreception for European Robin Cryptochrome 4. Curr. Biol. 2018, 28, 211–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancar, A. Structure and Function of DNA Photolyase and Cryptochrome Blue-Light Photoreceptors. Chem. Rev. 2003, 103, 2203–2238. [Google Scholar] [CrossRef]
- Sorigué, D.; Légeret, B.; Cuiné, S.; Blangy, S.; Moulin, S.; Billon, E.; Richaud, P.; Brugière, S.; Couté, Y.; Nurizzo, D.; et al. An algal photoenzyme converts fatty acids to hydrocarbons. Science 2017, 357, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Heuts, D.P.; Scrutton, N.S.; McIntire, W.S.; Fraaije, M.W. What’s in a covalent bond? On the role and formation of covalently bound flavin cofactors. FEBS J. 2009, 276, 3405–3427. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, P.A.; Feenstra, K.A.; Mark, A.E.; Berendsen, H.J.; Visser, A.J. Dynamic Conformations of Flavin Adenine Dinucleotide: Simulated Molecular Dynamics of the Flavin Cofactor Related to the Time-Resolved Fluorescence Characteristics. J. Phys. Chem. B 2002, 106, 8858–8869. [Google Scholar] [CrossRef]
- Kao, Y.-T.; Saxena, C.; He, T.-F.; Guo, L.; Wang, L.; Sancar, A.; Zhong, D. Ultrafast Dynamics of Flavins in Five Redox States. J. Am. Chem. Soc. 2008, 130, 13132–13139. [Google Scholar] [CrossRef] [Green Version]
- Stanley, R.; McFarlane, M. Ultrafast Excited State Dynamics of Oxidized Flavins: Direct Observations of Quenching by Purines. J. Phys. Chem. A 2000, 104, 6899–6906. [Google Scholar] [CrossRef]
- Spexard, M.; Immeln, D.; Thöing, C.; Kottke, T. Infrared spectrum and absorption coefficient of the cofactor flavin in water. Vib. Spectrosc. 2011, 57, 282–287. [Google Scholar] [CrossRef]
- Li, G.; Glusac, G.D. Light-Triggered Proton and Electron Transfer in Flavin Cofactors. J. Phys. Chem. B Lett. 2009, 113, 9059–9061. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Sichula, V.; Glusac, K.D. Role of Adenine in Thymine-Dimer Repair by Reduced Flavin-Adenine Dinucleotide. J. Phys. Chem. B 2008, 112, 10758–10764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, A.; Khade, R.V.; Hazra, P. pH dependent dynamic behavior of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) in femtosecond to nanosecond time scale. J. Photochem. Photobiol. A Chem. 2011, 221, 105–112. [Google Scholar] [CrossRef]
- Bubniene, U.; Mazetyte, R.; Ramanaviciene, A.; Gulbinas, V.; Ramanavicius, A.; Karpicz, R. Fluorescence Quenching-Based Evaluation of Glucose Oxidase Composite with Conducting Polymer, Polypyrrole. J. Phys. Chem. C 2018, 122, 9491–9498. [Google Scholar] [CrossRef]
- Weigel, A.; Dobryakov, A.; Klaumünzer, B.; Sajadi, M.; Saalfrank, P.; Ernsting, N.P. Femtosecond stimulated Raman spectroscopy of flavin after optical excitation. J. Phys. Chem. B 2011, 115, 3656–3680. [Google Scholar] [CrossRef]
- Rieff, B.; Mathias, G.; Bauer, S.; Tavan, S. Density Functional Theory Combined with Molecular Mechanics: The Infrared Spectra of Flavin in Solution. Photochem. Photobiol. 2011, 87, 511–523. [Google Scholar] [CrossRef]
- Wolf, M.M.N.; Schumann, C.; Gross, R.; Domratcheva, T.; Diller, R. Ultrafast Infrared Spectroscopy of Riboflavin: Dynamics, Electronic Structure, and Vibrational Mode Analysis. J. Phys. Chem. B 2008, 112, 13424–13432. [Google Scholar] [CrossRef]
- Neiss, C.; Saalfrank, P.; Parac, M.; Grimme, S. Quantum Chemical Calculation of Excited States of Flavin-Related Molecules. J. Phys. Chem. A 2003, 107, 140–147. [Google Scholar] [CrossRef]
- HyperChem(TM) Professional 7.51; Hypercube, Inc.: Gainesville, FL, USA, 2003.
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–224. [Google Scholar]
- Visser, A.J.W.G. Kinetics of Stacking Interactions in Flavin Adenine Dinucleotide from Time Resolved Flavin Fluorescence. Photochem. Photobiol. 1984, 40, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ehrmaier, J.; Sobolewski, A.L.; Karsili, T.N.V.; Domcke, W. Mechanisms of photoreactivity in hydrogen-bonded adenine–H2O complexes. Phys. Chem. Chem. Phys. 2019, 21, 14238–14249. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Singlet | Triplet | Diff (Triplet−Singlet) | |
|---|---|---|---|
| Structure 1 | −3885.38051400 a.u. | −3885.30052427 a.u. | 50.19 kcal |
| Structure 2 | −3885.37366640 a.u. | −3885.31405627 a.u. | 37.41 kcal |
| Diff (Struc1−Struc2) | −4.29 kcal | −8.49 kcal |
flavin ring: N1, C10a, C4a, N5  | flavin C4a – C10a  | flavin C4a – N5  | flavin C10a-N1  |
| FAD triplet | 1.40 Å | 1.35 Å | 1.32 Å |
| FAD singlet | 1.44 Å | 1.30 Å | 1.30 Å |
| FAD without water clip | 1.44 Å | 1.31 Å | 1.29 Å |
| Structure 1 | Structure 2 | |||
|---|---|---|---|---|
| MO | Singlet [a.u.] | Triplet [a.u.] | Singlet [a.u.] | Triplet [a.u.] |
| 238 | −0.2473 | −0.2511 −0.2383 | ||
| 239 | −0.2360 HOMO | −0.2399 −0.1785 HOMO | −0.2356 HOMO | −0.2398 −0.1785 |
| 240 | −0.1059 LUMO | −0.1722 LUMO −0.0854 | −0.1188 LUMO | −0.1722 −0.0852 HOMO |
| 241 | −0.0417 | −0.0454 LUMO −0.0329 | ||
| Open FAD (gas phase) | [scaled] | Open FAD (Microsolvation) | [scaled] | FAD in Structure 1 | [scaled] | Experiment | |
|---|---|---|---|---|---|---|---|
| C4=O | 1830 [1757] | C4=O, water | 1835 [1762] | C4=O, water | 1804 [1732] | C4=O | 1712 |
| C2=O and H-bridge amine (adenine) | 1774 [1703] | C2=O, water | 1783 [1712] | C2=O, water | 1758 [1687] | C2=O | 1657 |
| Adenine | 1668 [1600] | Adenine | 1700 [1632] | adenine, water clip 1 | 1680 [1612] | Adenosine | 1657 |
| ring I, II, III, amine (adenine) | 1638 [1572] | ring I, II, III, amine (adenine) | 1640 [1574] | ring I, II, III, water clip 1 & 2, solvating water | 1640 [1574] | band I | 1626 |
| Adenosine | 1605 | ||||||
| ring I, II, III, amine (adenine) | 1591 [1527] | ring I, II, III, amine (adenine), solvating water | 1594 [1530] | ring I, II, III, water clip 3 | 1594 [1530] | band II | 1580 |
| band III | 1547 | ||||||
| band IV | 1506 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kieninger, M.; Ventura, O.N.; Kottke, T. Calculation of the Geometries and Infrared Spectra of the Stacked Cofactor Flavin Adenine Dinucleotide (FAD) as the Prerequisite for Studies of Light-Triggered Proton and Electron Transfer. Biomolecules 2020, 10, 573. https://doi.org/10.3390/biom10040573
Kieninger M, Ventura ON, Kottke T. Calculation of the Geometries and Infrared Spectra of the Stacked Cofactor Flavin Adenine Dinucleotide (FAD) as the Prerequisite for Studies of Light-Triggered Proton and Electron Transfer. Biomolecules. 2020; 10(4):573. https://doi.org/10.3390/biom10040573
Chicago/Turabian StyleKieninger, Martina, Oscar N. Ventura, and Tilman Kottke. 2020. "Calculation of the Geometries and Infrared Spectra of the Stacked Cofactor Flavin Adenine Dinucleotide (FAD) as the Prerequisite for Studies of Light-Triggered Proton and Electron Transfer" Biomolecules 10, no. 4: 573. https://doi.org/10.3390/biom10040573
APA StyleKieninger, M., Ventura, O. N., & Kottke, T. (2020). Calculation of the Geometries and Infrared Spectra of the Stacked Cofactor Flavin Adenine Dinucleotide (FAD) as the Prerequisite for Studies of Light-Triggered Proton and Electron Transfer. Biomolecules, 10(4), 573. https://doi.org/10.3390/biom10040573