1. Introduction
Acute kidney injury (AKI) is a clinical syndrome characterized by a rapid decline in kidney function and failure to regulate fluid, electrolyte, and acid–base balance. AKI originates from a variety of causes, including renal hypoperfusion (due to low cardiac output or reduced renal perfusion pressure), obstruction of the urinary tract, infections and sepsis, and exposure to nephrotoxins [
1,
2]. A common mechanism for AKI is ischemia (inadequate O
2 and nutrient delivery)-reperfusion injury, which involves mitochondrial dysfunction, oxidative stress, insufficient production of ATP, renal cell apoptosis and necrosis, and inflammation [
1,
2,
3]. Mitochondrial dysfunction and ATP deficits are the most pronounced in tubular cells of renal cortex and precede the clinical manifestations of AKI [
2,
3,
4,
5]. As our understanding of the complex mechanisms involved in mitochondrial dysfunction remains inadequate, the therapies that reduce mitochondrial damage and/or stimulate mitochondrial biogenesis in the injured kidney are very limited.
An efficient exchange of solutes and nucleotides between mitochondria and the cytoplasm is required to maintain cellular energy metabolism. Among the three identified subtypes of the voltage dependent anion channel (VDAC1, VDAC2, and VDAC3), VDAC1 is most widely expressed and present in the majority of cell types [
6,
7,
8,
9,
10,
11]. VDAC1 is the major gateway for solute transport between the cytoplasm and mitochondria. VDAC1 is the most abundant integral protein component of the outer mitochondrial membrane and, in physiological conditions, mediates the bidirectional transport of ions, nucleotides (e.g., NAD
+/NADH, ADP/ATP), and solutes smaller than 5 kDa in size [
6]. Thus, VDAC1 is a crucial regulator of mitochondrial energy metabolism and Ca
2+ homeostasis [
7,
8,
9]. However, under pathophysiologic conditions, VDAC1 is involved in apoptosis; both at the early stages (Ca
2+ influx into mitochondria) as well as in later stages, when pro-apoptotic proteins are released from the mitochondria, and in ferroptosis [
8,
12,
13,
14,
15]. Apoptosis is associated with increased expression of VDAC1 in the outer mitochondrial membrane and oligomerization of VDAC1 monomers to form large channels that allow for release of mitochondrial pro-apoptotic proteins into the cytoplasm where they mediate additional apoptotic events [
10,
15,
16,
17]. Regulation of cell survival signals by VDAC1 occurs through its interactions with proteins of the Bcl-2 family, hexokinase, protein kinases, cytoskeletal proteins (tubulin, α-synuclein, plectin, and desmin), and mitochondrial lipids (cardiolipin and phoshatidylethanolamine) [
16,
17,
18,
19,
20,
21,
22,
23,
24].
VDAC1 closure results in the decrease or loss of the outer mitochondrial membrane permeability and accumulation of metabolites, nucleotides, and protons in the intermembrane space. If this condition persists, the outer membrane integrity is lost, mitochondrial homeostasis is disrupted, and cytochrome c diffuses to the cytosol thereby initiating apoptosis [
25]. In contrast, VDAC1 opening prevents events leading to apoptosis [
19,
26]. Thus, VDAC1 permeability has a critical impact not only on mitochondrial homeostasis, metabolic functions, and stress, but also on cellular ATP levels, injury, and fate.
In contrast to a substantial amount of knowledge of the role of VDAC1 in physiology and pathology of neuronal, cardiac, and cancer cells, very little is known about the role of VDAC1 in kidney physiology, injury, and regeneration. GSK3β-mediated phosphorylation of a threonine residue(s) of VDAC1 leads to mitochondrial permeability transition and death in cultured renal epithelial tubular cells [
27]. Inhibition of GSK3β blocks phosphorylation of VDAC1, attenuates mitochondrial permeability transition, reduces mitochondrial dysfunction and oxidative stress, and ameliorates oxidant-induced AKI [
27]. Deletion of VDAC3 increases mitochondrial production of the reactive oxygen species, alters renal sodium transport, and leads to hypertension whereas deletion of VDAC2 is lethal during embryonic development [
28,
29]. Oligomerization of VDAC1 has been implicated in cisplatin-induced apoptosis and nephrotoxicity due to a formation of pores in the outer mitochondrial membrane that allow for the escape of cytochrome c from the mitochondrial intermembrane space to the cytosol [
30]. As VDAC1 conductance can differentially mitigate or exacerbate pathological conditions in different organs and cell types, the purpose of this study was to determine the role of VDAC1 in (1) ischemia-induced mitochondrial dysfunction and injury to the kidney, and (2) kidney repair after ischemia.
3. Results
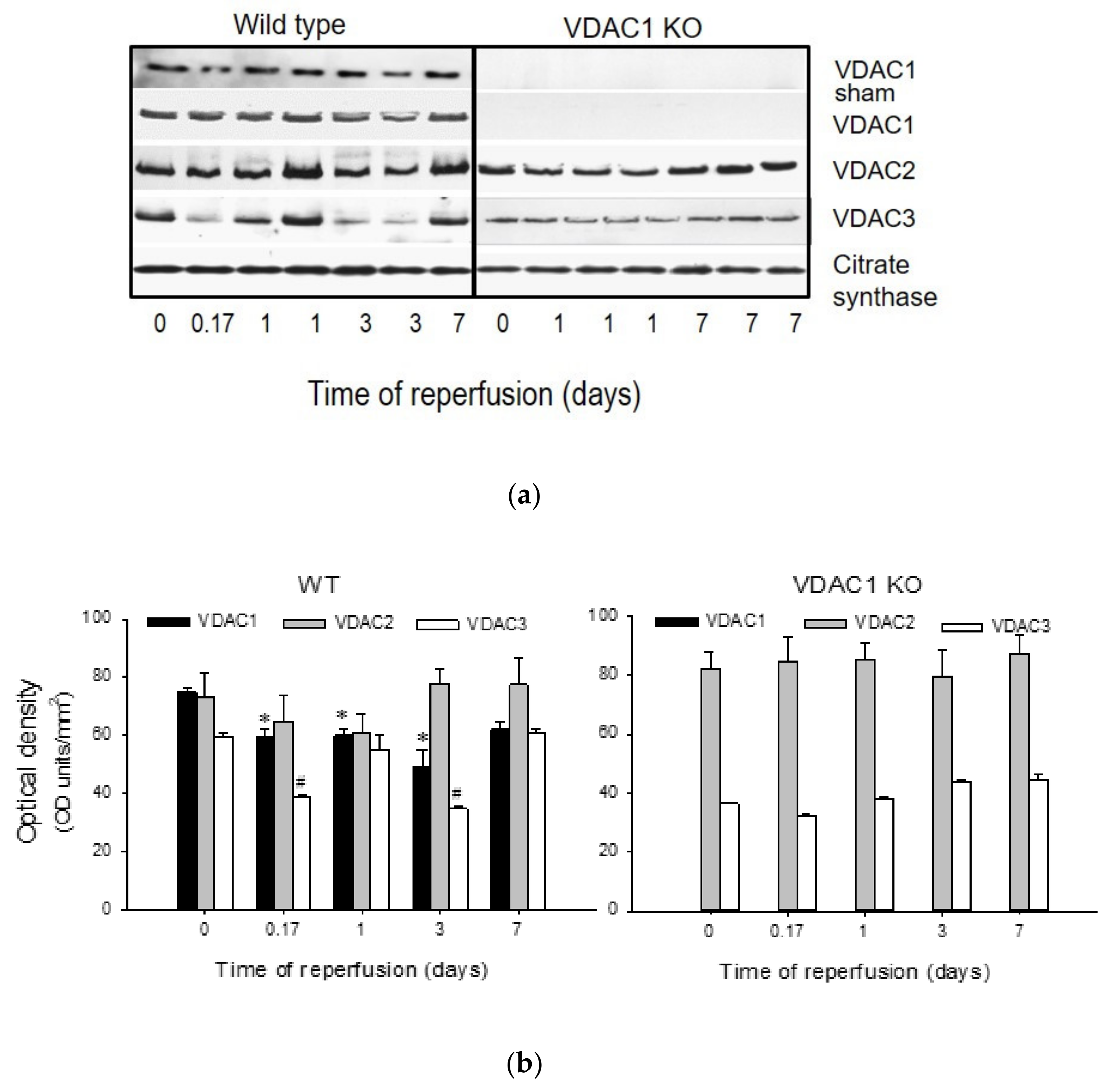
3.1. Ischemia Decreases the Levels of VDAC1 in Renal Cortical Mitochondria
Protein levels of VDAC1, VDAC2, and VDAC3 were assessed in mitochondria isolated from renal cortex of sham-operated and ischemic WT and VDAC1 KO mice at different times post reperfusion. Mitochondrial VDAC1 levels did not change in WT sham animals, but decreased after ischemia with the lowest level found on day 3 post reperfusion. On day 7 after ischemia, VDAC1 levels were not different from sham controls (
Figure 1). Likewise, mitochondrial levels of VDAC2 decreased after ischemia and returned to the control levels by day 7 (
Figure 1). Mitochondrial levels of VDAC3 decreased shortly (4 h) after ischemia, remained decreased until day 3 post reperfusion, and recovered by day 7 (
Figure 1). As shown in
Figure 1, VDAC1 protein was absent in mitochondria from VDAC1 KO mice. VDAC1 deletion did not alter the changes in renal mitochondrial levels of VDAC2 and VDAC3 in response to ischemia (
Figure 1).
These data show that ischemia decreases the levels of all known VDAC isoforms and that the kidney recovery after ischemic injury is associated with the return of mitochondrial levels of VDAC isoforms in the kidneys of wild type mice.
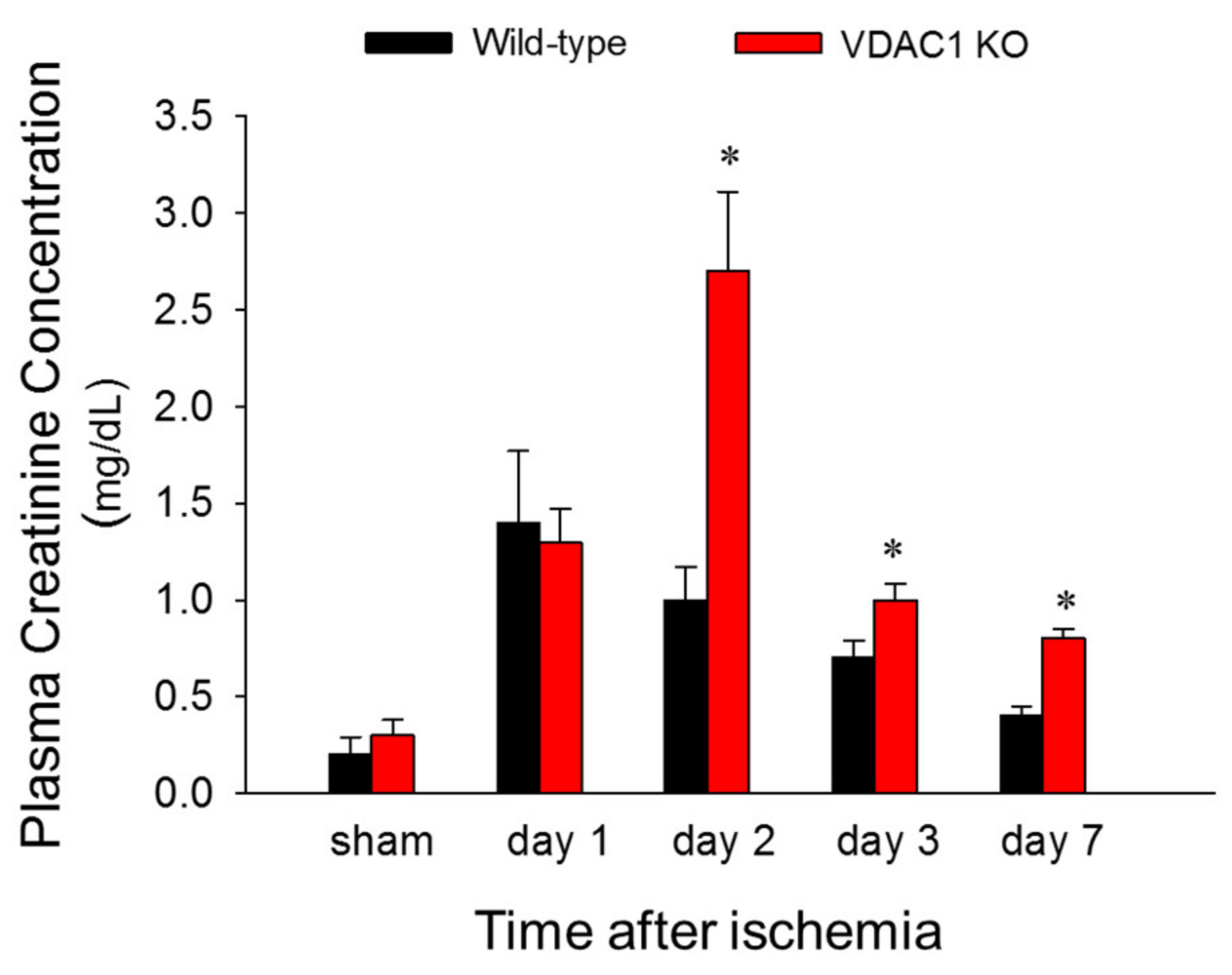
3.2. Deficiency of VDAC1 Delays Recovery of Renal Function After Ischemia
To test if VDAC1 is involved in changes in renal function induced by ischemia, blood samples were taken at different times post reperfusion and the levels of plasma creatinine (a marker of renal function) were assessed. There was no difference in plasma creatinine levels between WT and VDAC1 KO sham mice. Creatinine levels increased 6-fold and 5-fold at 24 and 48 h after reperfusion, respectively, and recovered 7 days after ischemia in WT mice (
Figure 2). VDAC1 deletion had no effect on the creatinine levels in sham mice (
Figure 2). Creatinine levels increased 6-fold at 24 h after ischemia in VDAC1 KO mice and these increases were equivalent WT and VDAC KO animals (
Figure 2). However, in contrast to WT mice, creatinine levels in VDAC1 KO mice continued to increase until 48 h post reperfusion when they were 2.7-fold higher than in WT mice and 10-fold higher than creatinine levels in VDAC1 KO sham controls (
Figure 2). In contrast to WT mice, creatinine levels did not recover on day 7 after ischemia in VDAC1 KO mice, and were 3-fold higher than those prior to ischemia (
Figure 2).
These data show that deletion of VDAC1 prolongs and exacerbates ischemia-induced decline in renal function and delays recovery of renal function after AKI.
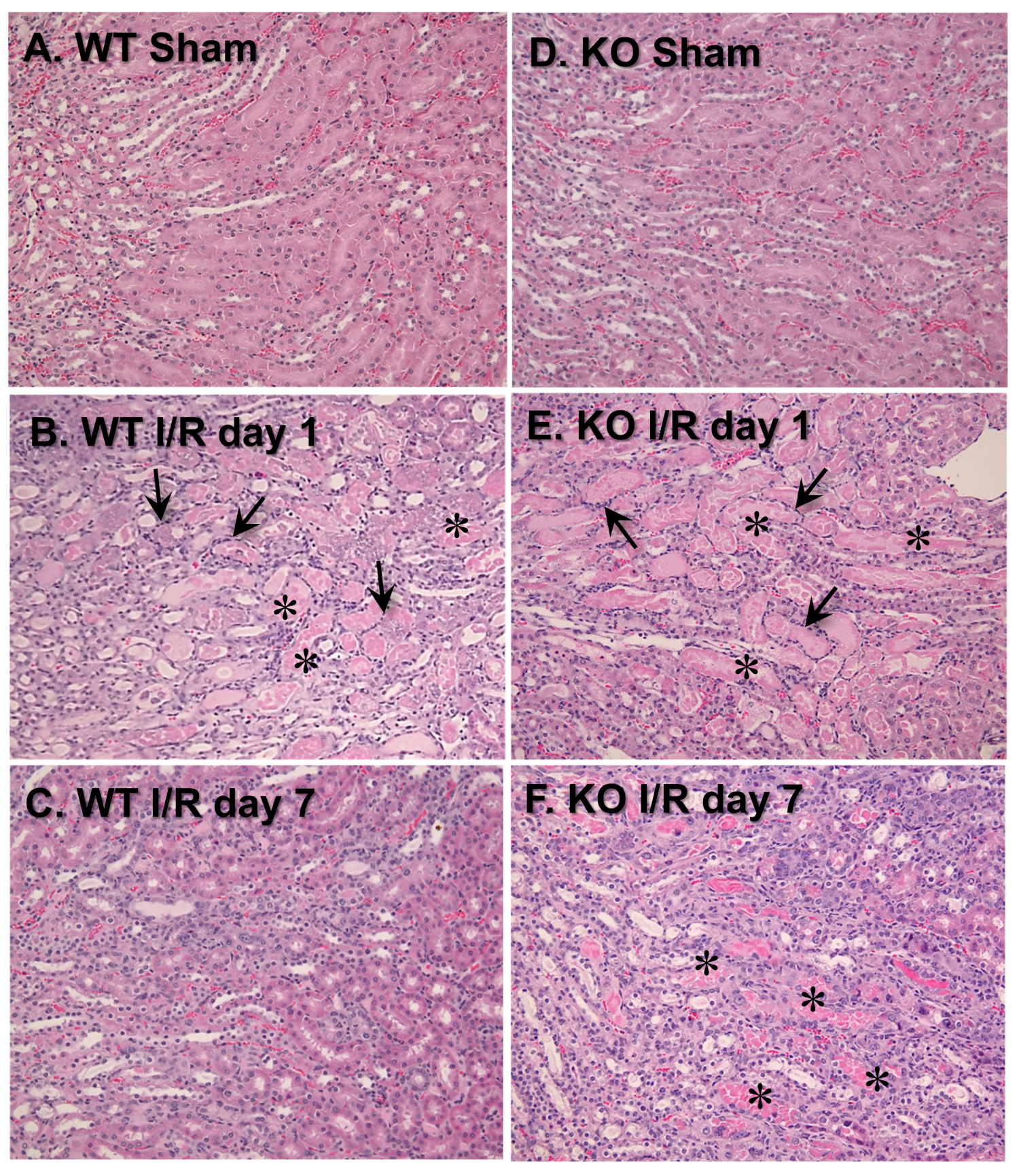
3.3. VDAC1 Deletion Impedes the Recovery of Kidney Morphology After Ischemia
To test the involvement of VDAC1 in morphological changes in ischemic kidneys, different histologic criteria were assessed on days 1 and 7 post reperfusion. Majority of morphological changes found in kidneys of WT mice at 24 h after ischemia occurred at the cortico-medullary junction (
Figure 3A,B;
Table 1). The changes involved severe tubular necrosis and loss of the brush border, tubular degeneration, and formation of copious tubular casts, significant damage to the distal nephron, and increased number of inflammatory cells (
Figure 3A,B;
Table 1). On day 7 post reperfusion, regeneration (relining) of damaged proximal tubules was nearly complete although some tubules did not have fully regenerated brush border and single tubular casts were scattered throughout the cortex in kidneys of WT mice (
Figure 3C;
Table 1).
VDAC1 deletion had no significant effect on the extent of morphologic damage to the proximal portion of the nephron and the number of inflammatory cells at 24 h after ischemia, but impaired recovery of proximal tubule morphology and tubular cast removal (
Figure 3B,E;
Table 1). In comparison with WT kidneys, regeneration of the proximal tubular part of the nephron in VDAC1-deficient kidneys was significantly reduced, necrotic foci and casts were apparent, and increased numbers of inflammatory cells were still present on day 7 post reperfusion (
Figure 3C,F;
Table 1).
Interestingly, the damage to the distal portion of the nephron was absent at 24 h and throughout the recovery period after ischemia in VDAC1-deficient kidneys (
Figure 3B,E;
Table 1). Thus, these data demonstrate that the absence of VDAC1 does not exacerbate morphological damage to the proximal tubular segment of the nephron, but impairs recovery of proximal tubule morphology after ischemia. These results suggest that VDAC1 plays an active role in the regeneration of the proximal segment of the nephron. In contrast, the absence of VDAC1 diminishes ischemic damage to the distal segment of the nephron.
3.4. The Absence of VDAC1 Promotes Interstitial Changes in Renal Tissue After Ischemia
Acute kidney injury is often followed by augmented accumulation of extracellular matrix proteins, which often results in interstitial fibrosis. Interstitial fibrosis has been implicated in the mechanisms of progression from AKI to the incident chronic kidney disease [
36]. We hypothesized that impaired recovery of kidney morphology and function after AKI in VDAC1 KO mice might be associated with increased deposition and accumulation of extracellular matrix proteins in the kidney interstitium. Thus, we assessed the effect of VDAC1 deficiency on collagen accumulation in the renal interstitium after ischemia using picro-sirius red staining. In comparison with the sham kidneys, positive red staining representing collagen presence in the interstitium was slightly increased in several areas of WT kidneys on day 7 after ischemia (
Figure 4A,B). VDAC1 deletion had no effect on collagen deposition in sham kidneys; however, it increased collagen accumulation in injured kidneys during the 7 day recovery period (
Figure 4D). These results show that the absence of VDAC1 increases accumulation of extracellular matrix proteins in the injured kidney and suggest that the lack of VDAC1 exacerbates ischemia-induced kidney fibrosis.
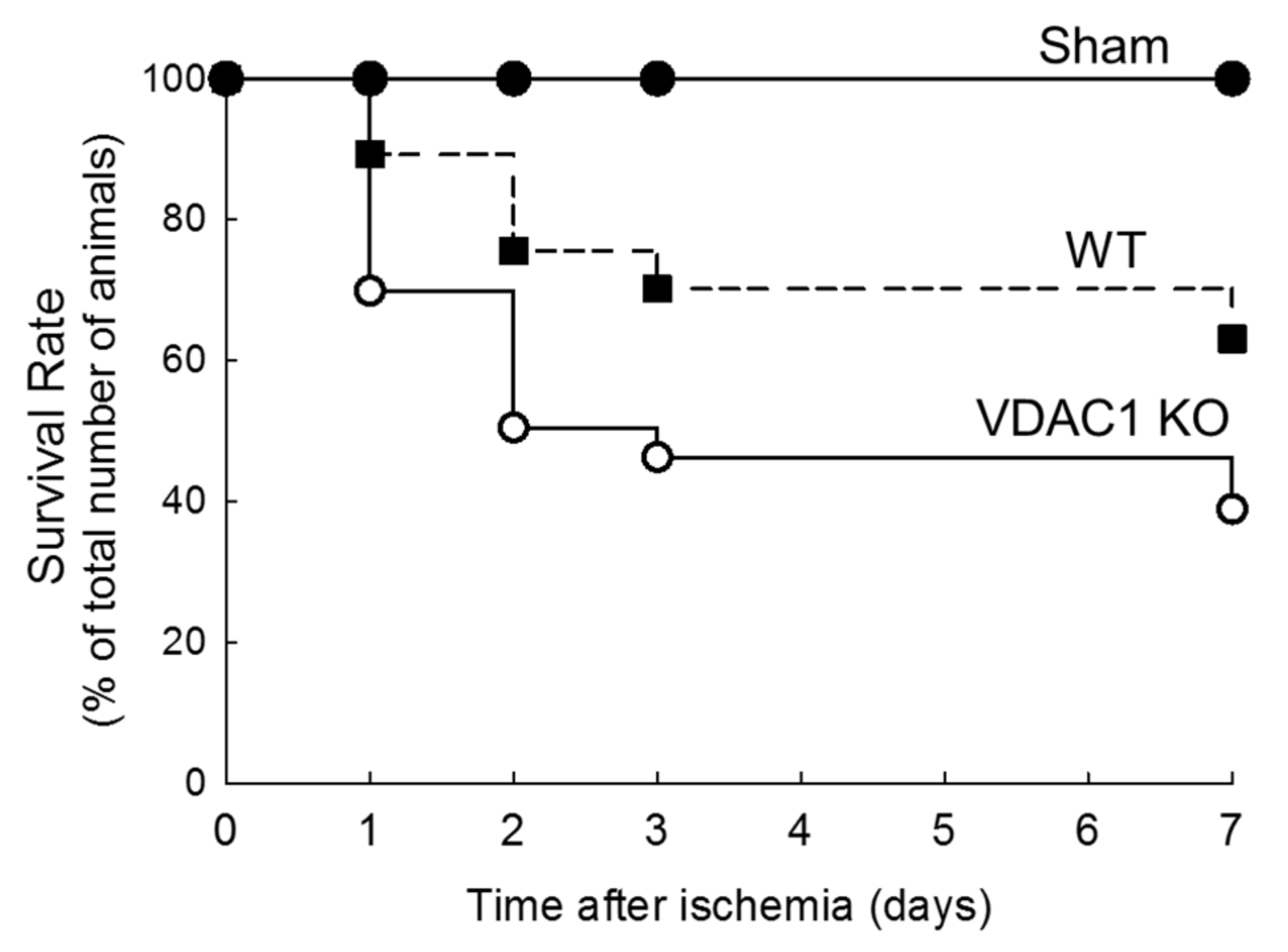
3.5. Deletion of VDAC1 Reduces Survival After Renal Ischemia
The survival rates were recorded to assess if deletion of VDAC1 and impaired recovery of kidney morphology and functions alter mice survival after ischemia.
Figure 5 shows that the highest mortality rate in both WT and VDAC1 KO mice occurred within 48 h after ischemia. The animal loss within this critical period was 24% in WT and 50% in VDAC1 KO mice. On the seventh day after ischemia, mortality rate was 37% and 61% in WT and VDAC1 KO mice, respectively (
Figure 5). No animals were lost from the sham-operated group. These results show that the deletion of VDAC1 increases (by 65%) mortality due to AKI and suggest that functional VDAC1 plays a critical role in survival after AKI (
Figure 5).
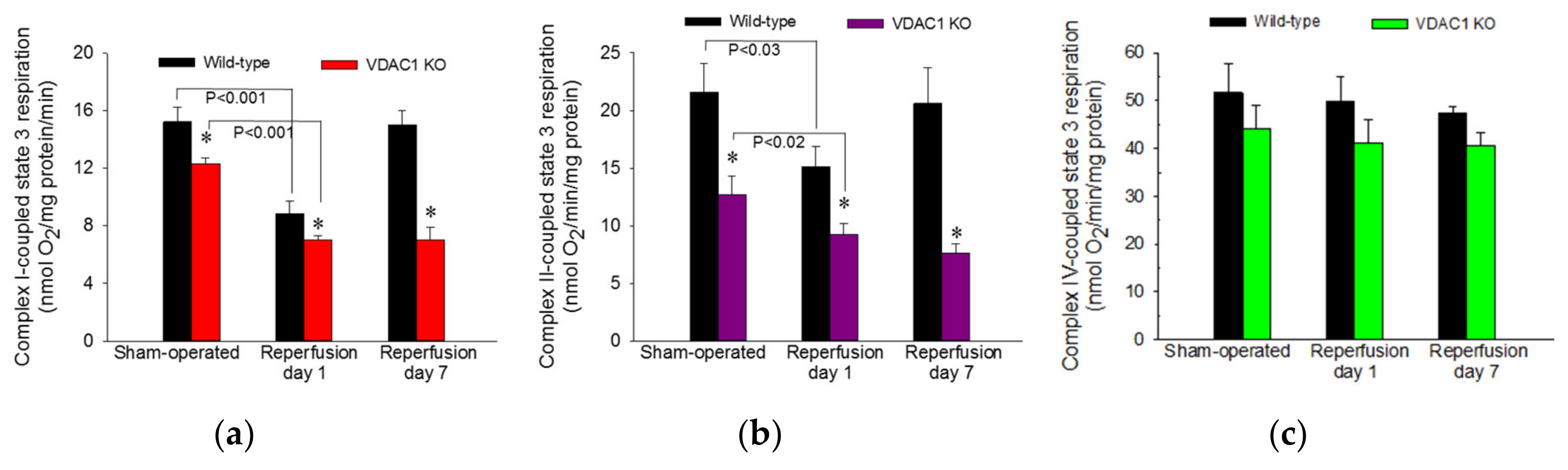
3.6. VDAC1 Deficiency Abrogates Recovery of Mitochondrial Respiration After Ischemia
The recovery of kidney morphology and functions after injury is an energy-consuming process that requires adequate ATP supply by mitochondria. Mitochondrial function is disrupted by ischemia and has to recover prior to the return of renal functions to meet the demands for ATP in the regenerating kidney. VDAC1 facilitates the movement of crucial solutes, substrates, and products of oxidative phosphorylation between the cytoplasm and mitochondria. Therefore, we examined the importance of this channel for the recovery of oxidative phosphorylation in the renal cortical mitochondria after ischemia-induced AKI.
ADP-stimulated state 3 respiration was used as a marker of the mitochondrial respiration associated with ADP phosphorylation and ATP synthesis in mitochondria isolated from renal cortical cells. State 3 respiration energized by glutamate and malate (substrates that generate NADH oxidized by complex I of the respiratory chain) was decreased to 58% of sham controls at 24 h after ischemia and recovered by day 7 after ischemia in WT mice (
Figure 6a). In non-injured kidneys, deletion of VDAC1 decreased state 3 respiration to 81% of that in WT mice (
Figure 6a). Ischemia in VDAC1 KO kidneys induced decreases in complex I-coupled state 3 respiration that were proportional to the decreases in WT kidneys (57% of sham controls at 24 h after reperfusion). However, in contrast to full recovery of respiration in WT kidneys, complex I-coupled state 3 respiration in VDAC1 KO renal cortical mitochondria did not recover and remained at the decreased level observed a day after ischemic injury (
Figure 6a).
State 3 respiration energized by the oxidation of succinate though complex II of the electron transport chain decreased to 70% of controls at 24 h and recovered within 7 days after ischemia in WT kidneys (
Figure 6b). Deletion of VDAC1 decreased complex II-coupled state 3 respiration in non-injured (sham) kidneys to 59% of that in WT sham mice. Ischemia produced further reduction in complex II-coupled state 3 respiration in VDAC1 KO animals. In contrast to full recovery of succinate-energized state 3 respiration in WT kidneys, state 3 respiration in VDAC1-deficient kidneys did not recover within 7 days post reperfusion and remained at the level observed at 24 h after ischemia (
Figure 6b). Neither ischemia nor deletion of VDAC1 had any significant effects on state 3 respiration energized by electron donors to complex IV (
Figure 6c).
These results demonstrate that VDAC1 deficiency decreases state 3 respiration in non-injured mitochondria and suggest that the access of substrates to mitochondrial matrix and the rate of electron flow through the respiratory chain are reduced in VDAC1 deficient kidneys. Moreover, the absence of VDAC1 blocks recovery of state 3 respiration after injury suggesting diminished capacity of VDAC1-deficient kidneys to produce ATP. Finally, these data show that VDAC1 is indispensable for efficient recovery of ADP-stimulated mitochondrial respiration following ischemia.
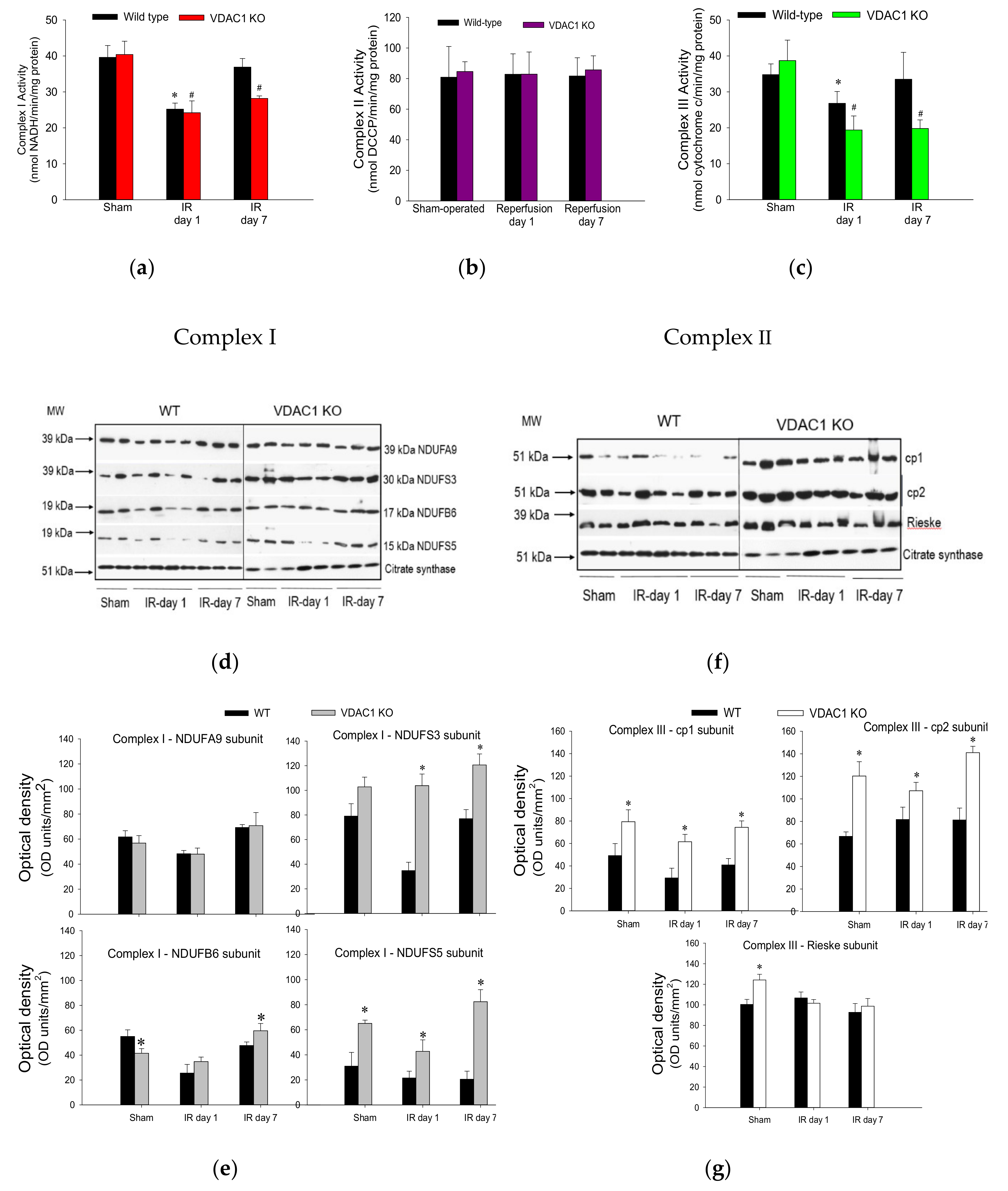
3.7. VDAC1 Deletion Hinders Recovery of Activities of Respiratory Complexes After Ischemia
To test whether decreases in state 3 respiration in VDAC1 KO mice are caused by a dysfunction of respiratory complexes, activities of complexes I, II, and III were assessed in renal mitochondria isolated from cortices of non-injured and ischemia-injured kidneys. The activity of complex IV was not measured because complex IV-coupled state 3 respiration was not altered in VDAC1-deficient kidneys.
Ischemia followed by 24 h reperfusion decreased activity of complex I in WT kidneys to 73% of the sham WT controls (
Figure 7a). This decrease was accompanied by a decline in protein levels of subunits NDUFA9 (39 kDa), NDUFS3 (30 kDa), NDUFB6 (17 kDa), and NDUFS5 (15 kDa) (
Figure 7d,e). The activity of complex I recovered by day 7 of post reperfusion, which was associated with the recovery of protein levels of its four subunits (
Figure 7a,d,e). Deletion of VDAC1 had no effect on the activity or protein levels of complex I in mitochondria isolated from non-injured kidneys (
Figure 7a,d,e). The ischemia-induced decreases in complex I activity were similar in WT and VDAC1-deficient kidneys at 24 h post-reperfusion (
Figure 7a). However, in contrast to the recovery of complex I activity after ischemia in WT kidneys, activity of this complex in VDAC1-deficient kidneys did not return and, on day 7 post reperfusion, it was 30% lower than that in respective sham controls (
Figure 7a). Interestingly, protein levels of NDUFA9, NDUFS3, and NDUFB6 subunits of complex I were not altered by ischemia or reperfusion in VDAC1-deficient kidneys, which suggests that the progressive decline and the lack of recovery of the activity of complex I after ischemia were not due to reduced levels of complex I in mitochondria (
Figure 7d,e).
Neither ischemia nor deletion of VDAC1 had any effect on the activity of complex II of the respiratory chain (
Figure 7b). These data suggest that the declines in mitochondrial state 3 respiration caused by ischemia and the deficiency of VDAC1 are not caused by insufficient protein levels of complex II in the mitochondria.
The activity of complex III in renal cortical mitochondria of WT mice declined to 61% of sham controls at 24 h after ischemia and recovered on day 7 of post reperfusion period (
Figure 7c). Deletion of VDAC1 had no effect on the activity of complex III in mitochondria isolated from non-injured kidneys but increased protein levels of subunits cp1, cp2, and Rieske iron-sulphur protein of complex III (
Figure 7c,f,g). Ischemia decreased the activity of complex III in VDAC1-deficient kidneys to 40% of respective sham controls without having a substantial effect on the protein levels of the major subunits of this complex (
Figure 7c,f,g). In addition, deletion of VDAC1 hindered recovery of complex III activity and, on day 7 post reperfusion, the activity of complex III was 30% lower than that in sham controls although protein levels of the major subunit of this complex were equivalent to those in sham kidneys and higher than in WT kidneys (
Figure 7c).
These results show that the lack of VDAC1 has a differential effect on the activities of complexes of the electron transport chain in mitochondria of non-injured, injured, and regenerating kidneys. The deficiency of VDAC1 has no effect on the activities of complexes I and II, and increases mitochondrial levels and activity of complex III. Moreover, the absence of VDAC1 blocks recovery of activities of complexes I and III after ischemia without reducing their protein levels. These results suggest that maintaining the conductance of VDAC1 is critical for the recovery of normal functions of the electron transport chain and mitochondrial respiration after ischemia and that the return of activities of complexes I and III is dependent on the functional VDAC1 channel. Our data also show that the absence of VDAC1 leads to elevated levels of important subunits of complex I and complex III, but these increases do not lead to augmented activity of these two complexes.
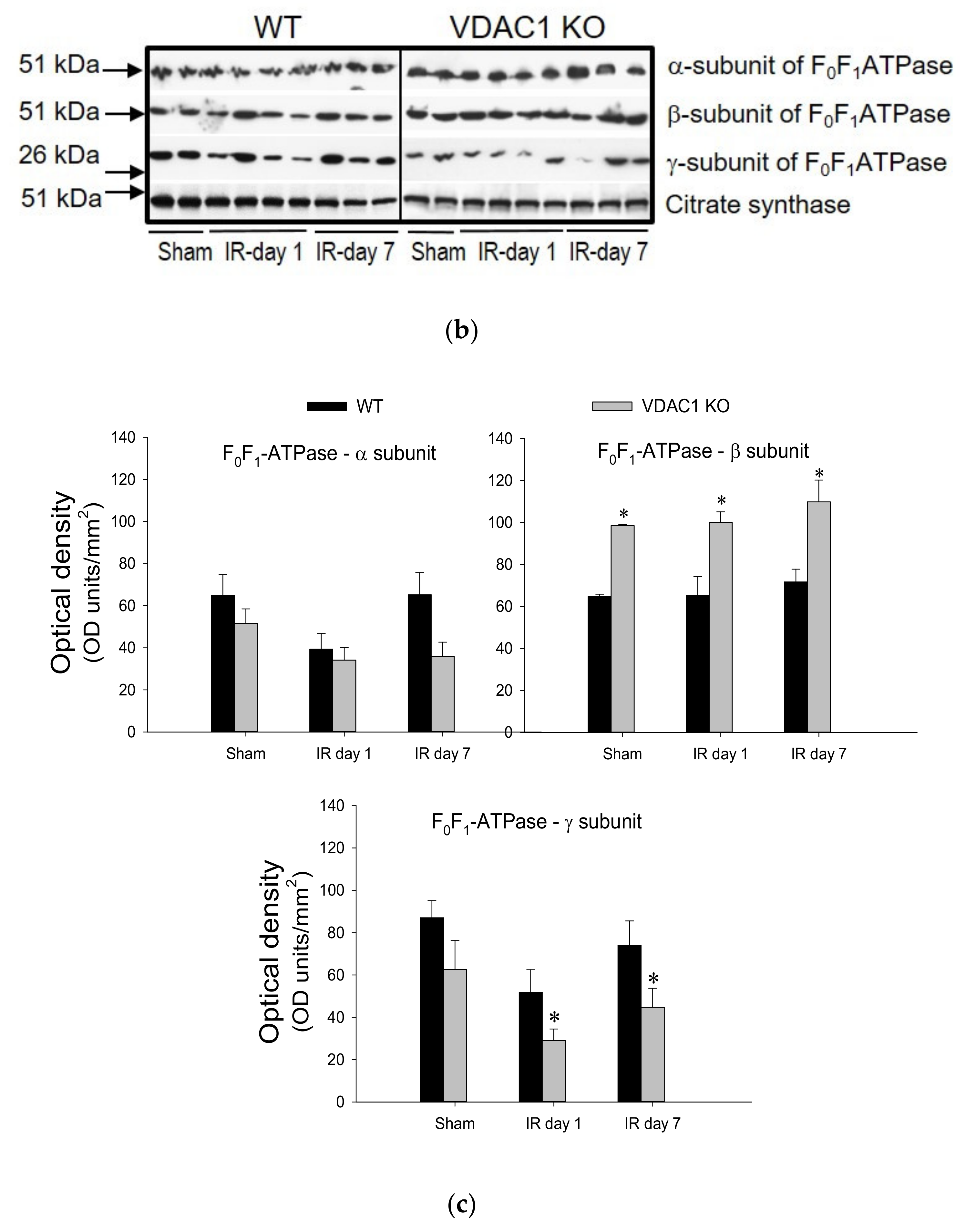
3.8. Deficiency of VDAC1 Impairs Recovery of FOF1-ATPase After Ischemia
This study also tested if VDAC1 plays a role in the decreases of F
0F
1-ATPase activity that occurs in AKI and recovery of F
0F
1-ATPase after AKI. Activity of F
0F
1-ATPase in mitochondria isolated from renal cortices of WT mice decreased to 65% of sham controls at 24 h after ischemia, was associated with the decreases in protein level of the γ-subunit of the enzyme, and recovered within 7 days post reperfusion (
Figure 8).
Deficiency of VDAC produced a 1.3-fold increase in F
0F
1-ATPase activity in non-injured kidneys and was accompanied by increased protein levels of the β (catalytic) subunit of the enzyme in renal cortical mitochondria (
Figure 8). Ischemia induced a decline in F
0F
1-ATPase activity in VDAC1-deficient kidneys similar to that in WT kidneys (
Figure 8). However, in contrast to the return of F
0F
1-ATPase activity after ischemia in WT kidneys, the F
0F
1-ATPase activity did not recover in VDAC1 KO kidneys and remained at the decreased level during the 7 days post reperfusion (
Figure 8). Lack of recovery of F
0F
1-ATPase activity after ischemia in VDAC1-deficient kidneys was accompanied by decreased mitochondrial levels of its γ-subunit (the central shaft connecting the F
o and F
1 subunits) in both non-injured and injured kidneys when compared to WT animals.
These data show that functional VDAC1 is essential for the full recovery of the catalytic activity of renal F0F1-ATPase after ischemia-induced AKI.
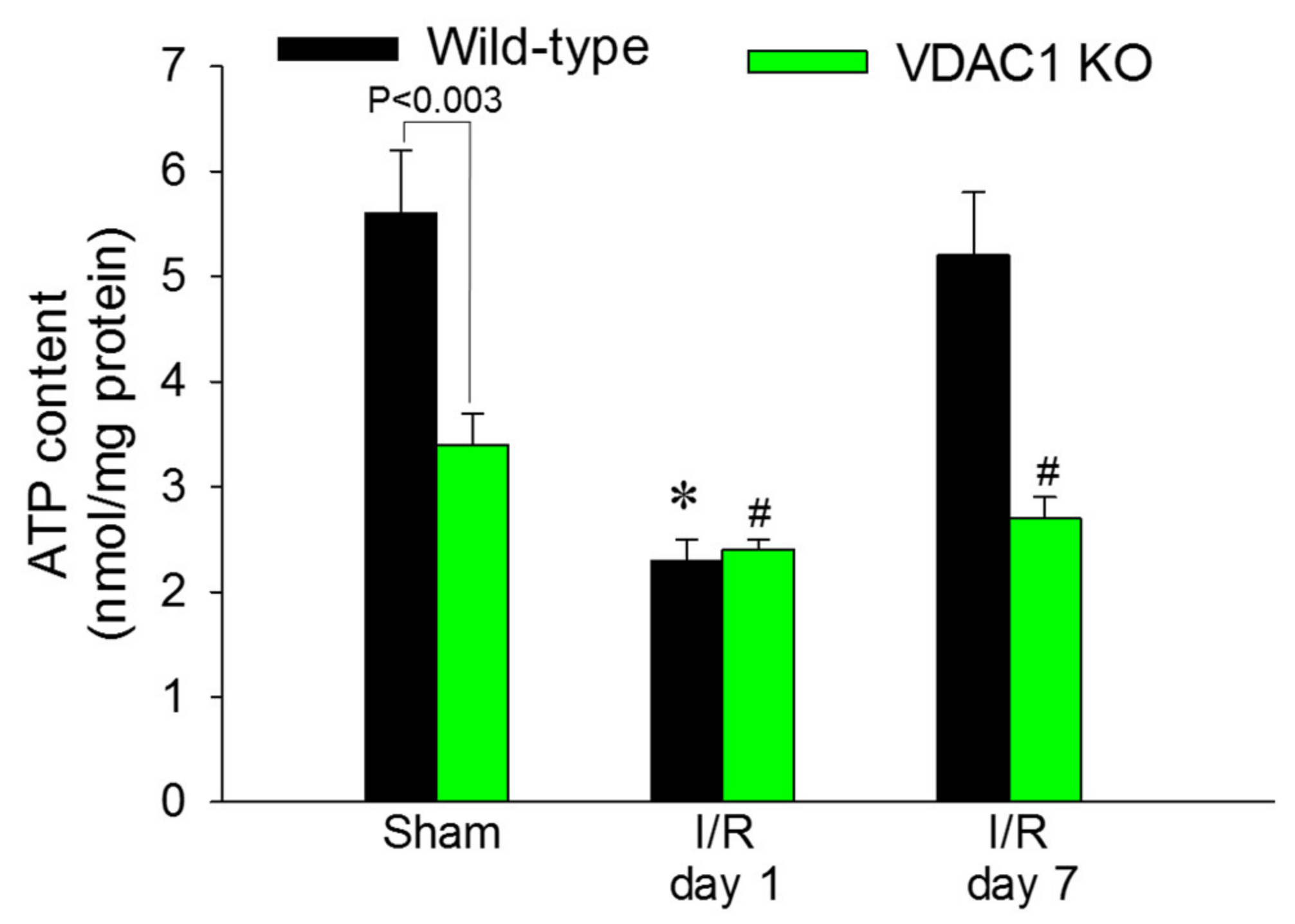
3.9. Deficiency of VDAC1 Reduces Renal Cortical ATP Content
ATP content in renal cortex was assessed to test whether decreases in state 3 respiration and F
0F
1-ATPase activity in VDAC1-deficient kidneys result in reduced levels of tissue ATP content. The slices of renal cortex were dissected immediately after the heart function ceased and were instantaneously denatured in ice-cold acid to prevent ATP consumption and hydrolysis. Renal cortical ATP levels in WT mice declined to 41% of sham controls at 24 h after reperfusion and recovered by day 7 after ischemia (
Figure 9).
VDAC1 deletion resulted in a 44% decrease in ATP content in cortices of non-injured kidneys in comparison with WT kidneys (
Figure 9). Ischemia reduced ATP content in kidneys of WT and VDAC1 KO mice to the same level (2.4 ± 0.1 vs. 2.3 ± 0.2 nmol/mg protein in renal cortical tissue of VDAC1 KO and WT mice, respectively). However, deletion of VDAC1 blocked the recovery of renal ATP levels after ischemia and on day 7 post reperfusion, ATP content in VDAC1-deficient kidneys has not reached the levels found in sham controls (
Figure 9). These data show that the presence of VDAC1 is crucial for maintaining ATP levels in the non-injured kidney. Furthermore, our data show that the lack of VDAC1 does not exacerbate ischemia-induced decreases in ATP, but blocks recovery of cellular ATP content after injury.
3.10. Lack of VDAC1 Is Associated with Mitochondrial Fission
Mitochondrial dysfunction is often associated with disruption of mitochondrial dynamics and increased fission (fragmentation) of this organelle. Drp1 is recruited to the mitochondria and binds to its corresponding receptors located on the mitochondrial outer membrane. Therefore, we used mitochondrial levels of DRP1 as a marker of the pool of fragmented mitochondria or mitochondria undergoing fission. No significant mitochondrial fission was present in non-injured kidneys of WT mice (
Figure 10a). Mitochondrial fission was observed after ischemia in WT kidneys and the fragmentation of mitochondria returned to undetectable levels on day 7 after injury (
Figure 10a).
In contrast, deficiency of VDAC1 was associated with increased levels of DRP1 in cortical mitochondria of non-injured kidneys, which suggested mitochondrial fragmentation in the absence of any injury (
Figure 10b). Ischemia augmented mitochondrial levels of DRP1 (
Figure 10b). These data show that ischemia induces association of DRP1 with renal cortical mitochondria suggesting alterations in mitochondrial dynamics and fragmentation. The lack of VDAC1 is sufficient to induce association of DRP1 with mitochondria in the absence of ischemic injury and promotes mitochondrial fission whereas ischemia augments this increase. These data suggest that VDAC1 plays a major role in the maintenance of mitochondrial dynamics in renal mitochondria.
4. Discussion
The VDAC1 channel is required for the movement of metabolites, including respiratory substrates, adenine nucleotides, Pi, and ions between the cytosol and mitochondria to maintain physiological functions of mitochondria [
42,
43]. Interestingly, changes in VDAC1 conductance do not affect creatine or phosphocreatine movements, which promote energy transfer through the phosphocreatine pathway [
44]. In addition to regulating mitochondrial energy metabolism, VDAC1 plays a role in cell injury and death, both necrotic and apoptotic cell death [
44,
45,
46]. Additionally, VDAC1 plays an important role in the release of mitochondrial reactive oxygen species that can initiate or exacerbate cell and organ injury [
28]. Consequently, changes in VDAC1 mitochondrial levels, oligomerization, and conductance can attenuate or exacerbate injury. It has been shown that diabetic nephropathy is accompanied by diminished levels of VDAC1 in the kidney [
27]; however, the role of individual VDAC isoforms in diabetic nephropathy or in mitochondrial dysfunction associated with acute kidney injury have not been assessed. Here, we demonstrate the presence of all three VDAC isoforms in the renal cortical cell mitochondria and examine whether VDAC1 plays a role in ischemia-induced dysfunction of renal mitochondria and subsequent acute injury caused by ischemic events in the kidney.
All three known VDAC isoforms are present in the renal cortical mitochondria and the functions of VDAC2 and VDAC3 may partially compensate for the lack of VDAC1 in non-injured kidneys to facilitate morphological and functional development of the kidney. However, in the absence of VDAC1, oxidative phosphorylation and tissue ATP levels are significantly reduced, which demonstrates that VDAC2 and/or VDAC3 do not fully substitute for the functions of VDAC1 in the non-injured kidney. Maximum (state 3, ADP-driven) mitochondrial respiration in non-injured kidneys of VDAC1 KO mice is reduced without any effect of VDAC1 deletion on the activities of complexes of the electron transport chain. A limited movement of metabolites and substrates/products of oxidative phosphorylation across the outer mitochondrial membrane in the absence of VDAC1 could explain this outcome. The transport of these solutes and ions across the outer mitochondrial membrane must occur through another gateway(s) and VDAC2 and/or VDAC3 could serve as these channels in non-injured kidney. However, this transport is reduced due to the absence of VDAC1 and is sufficient to support respiration and ATP synthesis, but at levels reduced in comparison with those in WT kidneys.
Cellular regeneration and recovery of cell functions after injury requires increased supply of ATP to support DNA repair, recycling of damaged macromolecules and organelles, repair of cellular membranes, and synthesis of new proteins and other macromolecules. Our data show that VDAC1 is a major protein necessary for the recovery of mitochondrial functions, ATP levels, and kidney morphology and functions after injury. Deletion of VDAC1 blocks recovery of ATP levels after ischemic injury, morphological regeneration of proximal tubules, and the return of kidney functions. In contrast, deletion of VDAC1 appears to prevent distal tubule damage. Distal tubules have higher expression of glycolytic enzymes than proximal tubules and are less dependent on mitochondria to produce ATP [
46]. Therefore, glycolysis can compensate for reduced oxidative phosphorylation when mitochondrial functions are compromised by ischemia. The absence of VDAC1 in proximal tubules does not exacerbate ischemia-induced decreases in state 3 respiration and the activities of complexes I and III, the two major targets of ischemia in the electron transport chain. However, deletion of VDAC1 blocks recovery of activities of these complexes following ischemia. This outcome was unexpected as protein levels of the major subunits of complex I and complex III in VDAC1-deficient injured kidneys were higher than in WT kidneys and the decreases in activities of these complexes could not be attributed to increased degradation or decreased synthesis of subunits of these two complexes. However, a substantial pool of these proteins was not functional after ischemia, which suggested that the subunits of complexes I and III were present, but their assembly was disrupted and could not be repaired in the absence of VDAC1. In contrast, the functions of complexes II and IV were unaffected by ischemia and lack of VDAC1. These data suggested that the hindered repair of complexes I and III, which have the major contribution to generation of the proton-motive force in mitochondria, blocked recovery of ATP production and content in injured kidneys lacking VDAC1. These results underscore the fact that VDAC1 is an important gateway for the entry of substrates and products of oxidative phosphorylation in injured proximal tubules and that the alternate channels do not allow for sufficient movement of these substrates/products during increased demand for energy in regenerating kidney. The lack of VDAC1 hinders recovery of ADP-driven respiration, ATP levels, morphology of the proximal tubules, and kidney functions after ischemic injury. These data support the conclusion that VDAC1 is an important VDAC isoform in renal cortical mitochondria and that it plays a major role in kidney recovery after ischemia-induced AKI.
Previously, we have shown that mitochondrial fragmentation (fission) in renal proximal tubular cells leads to reduced state 3 respiration, decreased activity of complexes I and III of the electron transport chain, hyperpolarization of the mitochondrial membrane, and reduced cellular ATP content [
41,
47]. Hence, we tested whether the absence of VDAC1 stimulates mitochondrial fission in non-injured and ischemia-injured mitochondria using the association of DRP1 with the mitochondria as a marker. DRP1 is an essential protein that mediates the final stage of mitochondrial fission by binding tightly to its specific receptors localized on the outer mitochondrial membrane. Wrapping of bound DRP1 molecules around the mitochondria leads to their fragmentation [
48]. DRP1 was absent from the mitochondria isolated from cortices of non-injured WT kidneys. In WT kidneys, ischemia disrupted normal mitochondrial dynamics and slightly increased association of DRP1 with the mitochondria indicating mitochondrial fission. These changes were associated with disruption of respiration and integrity of the electron transport chain, and decreased ATP content in the renal cortex. Recovery of mitochondrial function after ischemia in WT mice was accompanied by decreased association with DRP1, which suggested reduced mitochondrial fission. On day 7 post reperfusion, mitochondrial levels of DRP1 were almost undetectable by immunoblotting and were accompanied by the recovery of respiration, activities of complex I, complex III, and F
0F
1-ATPase, and tissue ATP content.
In contrast, non-injured mitochondria lacking VDAC1 exhibited significant levels of DRP1 that were higher than the levels of DRP1 in injured WT mitochondria. It is noteworthy that the decreases in state 3 respiration and ATP content coincided with mitochondrial fission in non-injured kidneys of VDAC1 KO mice. Ischemia exacerbated increases in mitochondrial levels of DRP1 in VDAC1 KO kidneys suggesting increased mitochondrial fission in comparison with WT kidneys. The disruption of mitochondrial dynamics in VDAC1-deficient kidneys was accompanied by the lack of recovery of state 3 respiration, activities of complex I, complex III, and F0F1-ATPase, and ATP levels after ischemia. These changes occurred despite increased protein levels of complex III subunits and unchanged levels of subunits of complex I. This suggests that mitochondrial fission and dysfunction are induced by the deficiency of VDAC1 and not by decreased protein levels of complexes of the electron transport chain and subunits of the catalytic domain of ATP synthase (F0F1-ATPase). Increases in protein levels of complex III and the catalytic (β) subunit of F0F1-ATPase in mitochondria lacking VDAC1 may be a compensatory response to decreased activity of these enzymatic complexes and reduced oxidative phosphorylation or due to decreased degradation of these proteins. Overall, these results show that VDAC1 plays an important role in the regulation of mitochondrial dynamics and that the deficiency of this channel induces mitochondrial fission and reductions in functions in non-injured kidneys, and prevents recovery of mitochondrial functions and normal mitochondrial dynamics after ischemic injury.
In summary, our results show that ischemia decreases protein levels of VDAC1 in the renal cortical mitochondria and that VDAC1 levels return during the recovery period in WT mice. These decreases coincide with reduced mitochondrial and renal functions and ATP levels, increases in mitochondrial fission, morphological damage to the proximal segment of the nephron, and interstitial accumulation of collagen. The return of protein levels of VDAC1 after ischemia is accompanied by recovery of mitochondrial dynamics and functions, morphological regeneration of the kidney, and the return of normal kidney functions. Deletion of VDAC1 results in reduced mitochondrial respiration and ATP levels and increased mitochondrial fission in non-injured kidneys without any apparent effect on renal morphology and functions. However, the absence of VDAC1 in injured kidneys hinders recovery of mitochondrial functions and dynamics, impedes regeneration of renal morphology and return of kidney functions, and increases deposition of collagen in renal interstitium after ischemia. Our results also suggest that in the absence of VDAC1, the other two isoforms i.e., VDAC2 and/or VDAC3, play a compensatory role in the kidney physiology and pathophysiology.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}