Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment



Abstract
:1. Introduction
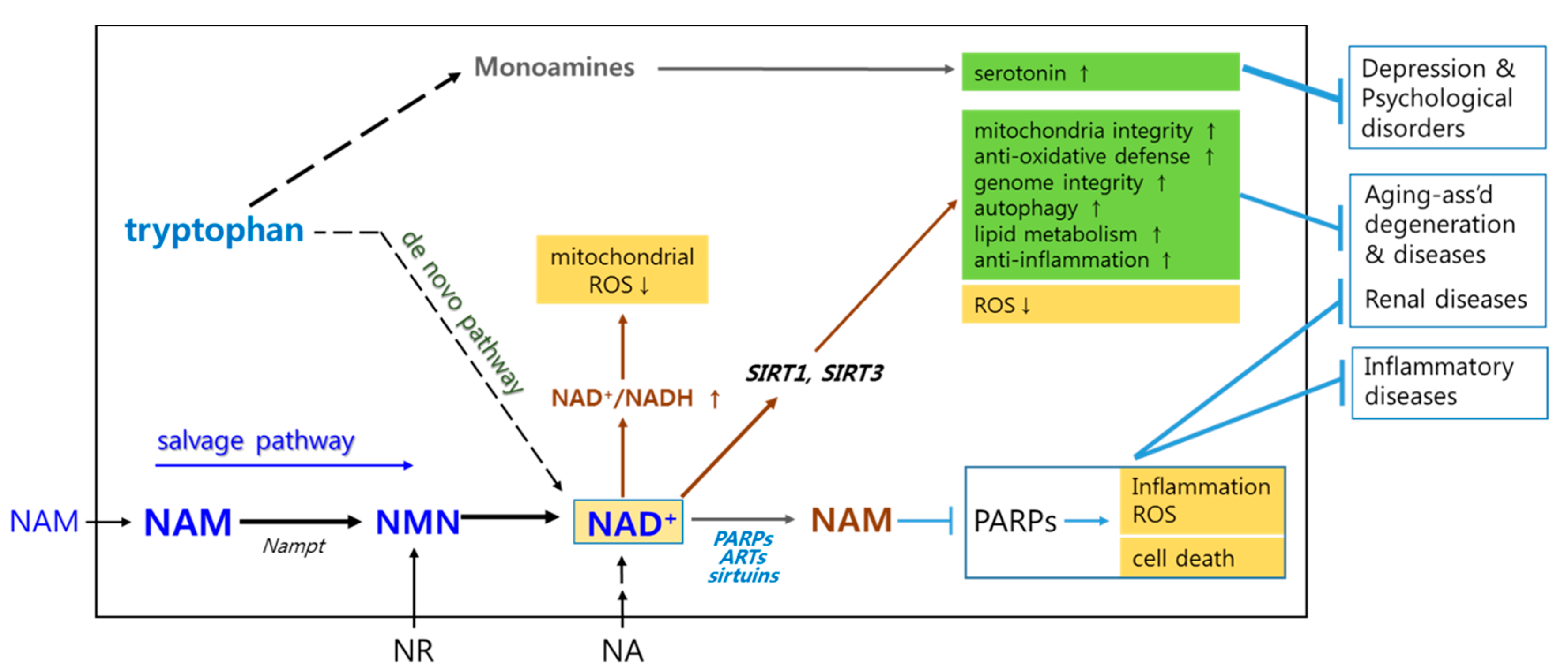
2. Briefs on Biochemistry Associated with Mechanisms Underlying NAM’s Positive Effects
3. Potential Toxicity and Adverse Effects of High Doses of NAM
3.1. Possible Genotoxicity and Carcinogenicity: Inconclusive Effects of NAM
3.2. Inhibition of Sirtuin Activity: An Effect that May Not Be Important In Vivo
3.3. High NAD+/NADH Ratio: Concerns Regarding Energy Metabolism
3.4. High-Level NAD+: Effect on Protein Translation
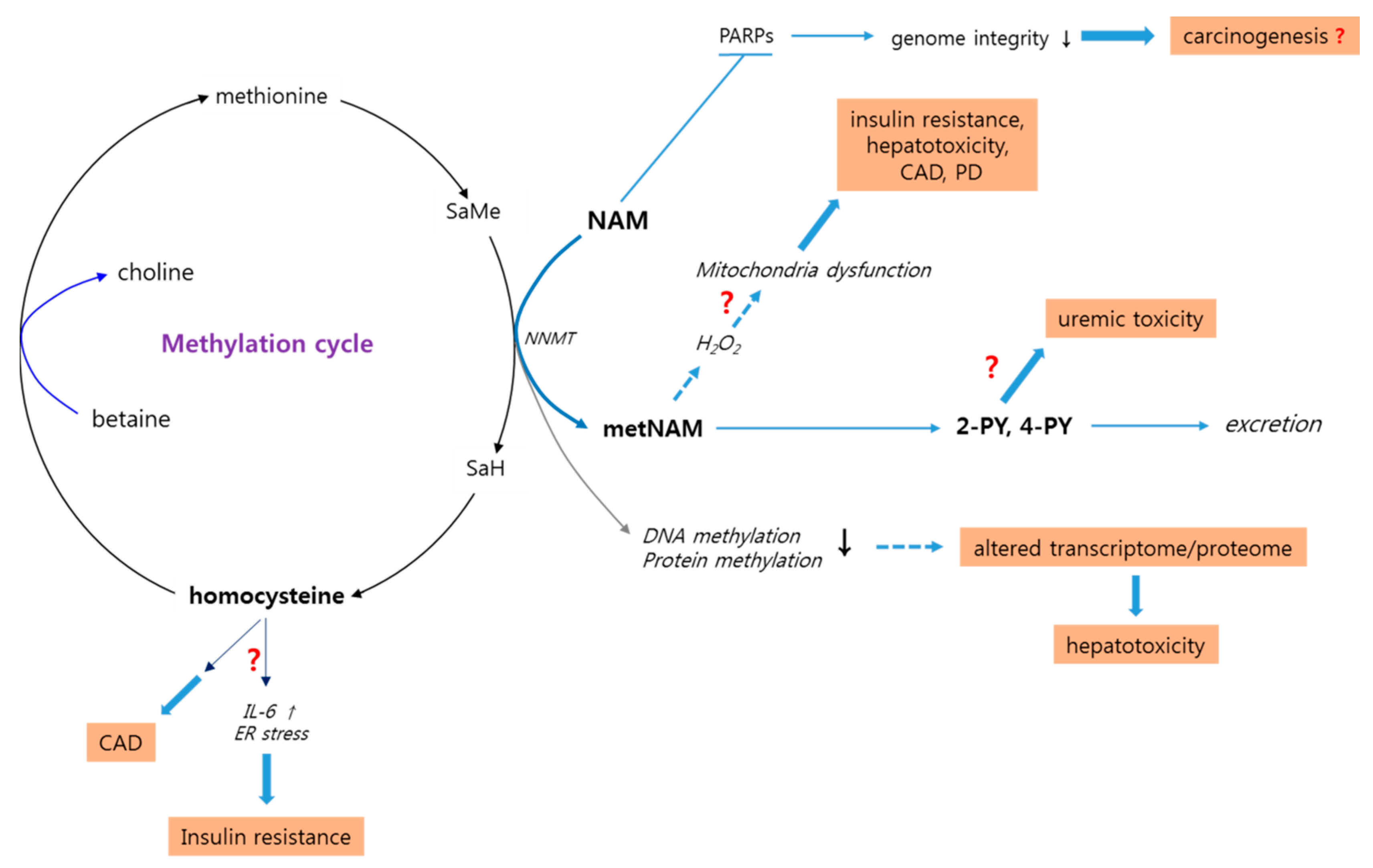
3.5. High-Level NAM Methylation: Potential Effects of Altered Methyl Pool
3.6. High-Level NAM Methylation: Potential Adverse Effects of Altered Methyl Pool
3.6.1. Insulin Resistance and Metabolic Syndrome
3.6.2. Parkinson’s Disease
3.6.3. Cardiac Diseases
3.6.4. Liver Toxicity
3.7. Potential Positive Effects of metNAM: Contradiction to the Proposed Adverse Effects
3.8. N-Methyl-2-Pyridone-5-Carboxamide: A Potential Uremic Toxin
4. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Elvehjem, C.A.; Madden, R.J.; Strong, F.M.; Wolley, D.W. The isolation and identification of the anti-black tongue factor. J. Biol. Chem. 1938, 123, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Klaidman, L.K.; Nalbandian, A.; Oliver, J.; Chang, M.L.; Chan, P.H.; Adams, J.D., Jr. The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in wistar rats. Neurosci. Lett. 2002, 333, 91–94. [Google Scholar] [CrossRef]
- Anderson, D.W.; Bradbury, K.A.; Schneider, J.S. Broad neuroprotective profile of nicotinamide in different mouse models of mptp-induced parkinsonism. Eur. J. Neurosci. 2008, 28, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Mokudai, T.; Ayoub, I.A.; Sakakibara, Y.; Lee, E.J.; Ogilvy, C.S.; Maynard, K.I. Delayed treatment with nicotinamide (vitamin b(3)) improves neurological outcome and reduces infarct volume after transient focal cerebral ischemia in wistar rats. Stroke 2000, 31, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Miao, C.Y. Nampt as a therapeutic target against stroke. Trends Pharmacol. Sci. 2015, 36, 891–905. [Google Scholar] [CrossRef]
- Sheline, C.T.; Cai, A.L.; Zhu, J.; Shi, C. Serum or target deprivation-induced neuronal death causes oxidative neuronal accumulation of zn2+ and loss of nad+. Eur. J. Neurosci. 2010, 32, 894–904. [Google Scholar] [CrossRef]
- Cai, A.L.; Zipfel, G.J.; Sheline, C.T. Zinc neurotoxicity is dependent on intracellular nad levels and the sirtuin pathway. Eur. J. Neurosci. 2006, 24, 2169–2176. [Google Scholar] [CrossRef]
- Hathorn, T.; Snyder-Keller, A.; Messer, A. Nicotinamide improves motor deficits and upregulates pgc-1alpha and bdnf gene expression in a mouse model of huntington’s disease. Neurobiol. Dis. 2011, 41, 43–50. [Google Scholar] [CrossRef]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laco, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in huntington’s disease models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of thr231-phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef]
- Bold, J.M.; Gardner, C.R.; Walker, R.J. Central effects of nicotinamide and inosine which are not mediated through benzodiazepine receptors. Br. J. Pharmacol. 1985, 84, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Hiromatsu, Y.; Sato, M.; Yamada, K.; Nonaka, K. Inhibitory effects of nicotinamide on recombinant human interferon-gamma-induced intercellular adhesion molecule-1 (icam-1) and hla-dr antigen expression on cultured human endothelial cells. Immunol. Lett. 1992, 31, 35–39. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Matsui, T.C.; Godin, A.M.; Gomides, L.F.; Pereira-Silva, P.E.; Duarte, I.D.; Menezes, G.B.; Coelho, M.M.; Klein, A. Neutrophil recruitment is inhibited by nicotinamide in experimental pleurisy in mice. Eur. J. Pharmacol. 2012, 685, 198–204. [Google Scholar] [CrossRef]
- Miesel, R.; Kurpisz, M.; Kroger, H. Modulation of inflammatory arthritis by inhibition of poly(adp ribose) polymerase. Inflammation 1995, 19, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Snaidr, V.A.; Damian, D.L.; Halliday, G.M. Nicotinamide for photoprotection and skin cancer chemoprevention: A review of efficacy and safety. Exp. Dermatol. 2019, 28 (Suppl. 1), 15–22. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.F. Nicotinamide: An oral antimicrobial agent with activity against both mycobacterium tuberculosis and human immunodeficiency virus. Clin. Infect. Dis. 2003, 36, 453–460. [Google Scholar] [CrossRef]
- Murray, M.F.; Srinivasan, A. Nicotinamide inhibits hiv-1 in both acute and chronic in vitro infection. Biochem. Biophys. Res. Commun. 1995, 210, 954–959. [Google Scholar] [CrossRef]
- Crino, A.; Schiaffini, R.; Manfrini, S.; Mesturino, C.; Visalli, N.; Beretta Anguissola, G.; Suraci, C.; Pitocco, D.; Spera, S.; Corbi, S.; et al. A randomized trial of nicotinamide and vitamin e in children with recent onset type 1 diabetes (imdiab ix). Eur. J. Endocrinol. 2004, 150, 719–724. [Google Scholar] [CrossRef]
- Elliott, R.B.; Pilcher, C.C.; Fergusson, D.M.; Stewart, A.W. A population based strategy to prevent insulin-dependent diabetes using nicotinamide. J. Pediatric Endocrinol. Metab. 1996, 9, 501–509. [Google Scholar] [CrossRef]
- Gale, E.A.; Bingley, P.J.; Emmett, C.L.; Collier, T.; European Nicotinamide Diabetes Intervention Trial Group. European nicotinamide diabetes intervention trial (endit): A randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 2004, 363, 925–931. [Google Scholar] [CrossRef]
- Staeva-Vieira, T.; Peakman, M.; von Herrath, M. Translational mini-review series on type 1 diabetes: Immune-based therapeutic approaches for type 1 diabetes. Clin. Exp. Immunol. 2007, 148, 17–31. [Google Scholar] [CrossRef]
- Maes, M.; Galecki, P.; Chang, Y.S.; Berk, M. A review on the oxidative and nitrosative stress (o&ns) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 676–692. [Google Scholar]
- Hollis, F.; van der Kooij, M.A.; Zanoletti, O.; Lozano, L.; Canto, C.; Sandi, C. Mitochondrial function in the brain links anxiety with social subordination. Proc. Natl. Acad. Sci. USA 2015, 112, 15486–15491. [Google Scholar] [CrossRef]
- Dedee, F.; Murrell, M.R.-Q. Management and Prognosis of Bullous Pemphigoid. 2019. Available online: https://www.uptodate.com/contents/management-and-prognosis-of-bullous-pemphigoid (accessed on 23 April 2020).
- Starr, P. Oral nicotinamide prevents common skin cancers in high-risk patients, reduces costs. Am. Health Drug. Benefits 2015, 8, 13–14. [Google Scholar] [PubMed]
- Damian, D.L.; Patterson, C.R.; Stapelberg, M.; Park, J.; Barnetson, R.S.; Halliday, G.M. Uv radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J. Investig. Dermatol. 2008, 128, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Park, J.S.; Chung, G.J.; Lee, I.H.; Hwang, E.S. Diverse therapeutic efficacies and more diverse mechanisms of nicotinamide. Metabolomics 2019, 15, 137. [Google Scholar] [CrossRef] [PubMed]
- Prousky, J. Vitamin b3 for depression: Case report and review of the literature. J. Orthomol. Med. 2010, 25, 137–147. [Google Scholar]
- Saini, J.S.; Corneo, B.; Miller, J.D.; Kiehl, T.R.; Wang, Q.; Boles, N.C.; Blenkinsop, T.A.; Stern, J.H.; Temple, S. Nicotinamide ameliorates disease phenotypes in a human ipsc model of age-related macular degeneration. Cell Stem Cell 2017, 20, 635-647.e7. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cardozo, B.H.; Cochran, K.E.; John, S.W.M. Nicotinamide and wld(s) act together to prevent neurodegeneration in glaucoma. Front. Neurosci. 2017, 11, 232. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; John, S.W.M. Glaucoma as a metabolic optic neuropathy: Making the case for nicotinamide treatment in glaucoma. J. Glaucoma 2017, 26, 1161–1168. [Google Scholar] [CrossRef]
- Nakajima, H.; Yamada, K.; Hanafusa, T.; Fujino-Kurihara, H.; Miyagawa, J.; Miyazaki, A.; Saitoh, R.; Minami, Y.; Kono, N.; Nonaka, K.; et al. Elevated antibody-dependent cell-mediated cytotoxicity and its inhibition by nicotinamide in the diabetic nod mouse. Immunol. Lett. 1986, 12, 91–94. [Google Scholar] [CrossRef]
- Monfrecola, G.; Gaudiello, F.; Cirillo, T.; Fabbrocini, G.; Balato, A.; Lembo, S. Nicotinamide downregulates gene expression of interleukin-6, interleukin-10, monocyte chemoattractant protein-1, and tumour necrosis factor-alpha gene expression in hacat keratinocytes after ultraviolet b irradiation. Clin. Exp. Dermatol. 2013, 38, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Nagai, A.; Matsumiya, H.; Hayashi, M.; Yasui, S.; Okamoto, H.; Konno, K. Effects of nicotinamide and niacin on bleomycin-induced acute injury and subsequent fibrosis in hamster lungs. Exp. Lung Res. 1994, 20, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Gurujeyalakshmi, G.; Iyer, S.N.; Hollinger, M.A.; Giri, S.N. Procollagen gene expression is down-regulated by taurine and niacin at the transcriptional level in the bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1996, 277, 1152–1157. [Google Scholar]
- Kim, S.K.; Yun, S.J.; Kim, J.; Lee, O.J.; Bae, S.C.; Kim, W.J. Identification of gene expression signature modulated by nicotinamide in a mouse bladder cancer model. PLoS ONE 2011, 6, e26131. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Lee, J.W.; Quan, C.; Youn, H.J.; Kim, H.M.; Bae, S.C. Nicotinamide inhibits growth of carcinogen induced mouse bladder tumor and human bladder tumor xenograft through up-regulation of runx3 and p300. J. Urol. 2011, 185, 2366–2375. [Google Scholar] [CrossRef]
- Omidian, M.; Khazanee, A.; Yaghoobi, R.; Ghorbani, A.R.; Pazyar, N.; Beladimousavi, S.S.; Ghadimi, M.; Mohebbipour, A.; Feily, A. Therapeutic effect of oral nicotinamide on refractory uremic pruritus: A randomized, double-blind study. Saudi J. Kidney Dis. Transplant. 2013, 24, 995–999. [Google Scholar]
- Pozzilli, P.; Visalli, N.; Buzzetti, R.; Cavallo, M.G.; Marietti, G.; Hawa, M.; Leslie, R.D. Metabolic and immune parameters at clinical onset of insulin-dependent diabetes: A population-based study. Imdiab study group. Immunotherapy diabetes. Metabolism 1998, 47, 1205–1210. [Google Scholar] [CrossRef]
- Kalckar, H.M.; Maxwell, E.S.; Strominger, J.L. Some properties of uridine diphosphoglucose dehydrogenase. Arch. Biochem. Biophys. 1956, 65, 2–10. [Google Scholar]
- Muller, D.; Mehling, H.; Otto, B.; Bergmann-Lips, R.; Luft, F.; Jordan, J.; Kettritz, R. Niacin lowers serum phosphate and increases hdl cholesterol in dialysis patients. Clin. J. Am. Soc. Nephrol. 2007, 2, 1249–1254. [Google Scholar] [CrossRef]
- Surjana, D.; Halliday, G.M.; Martin, A.J.; Moloney, F.J.; Damian, D.L. Oral nicotinamide reduces actinic keratoses in phase ii double-blinded randomized controlled trials. J. Investig. Dermatol. 2012, 132, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Hoyer, M.; Honess, D.J.; Dennis, I.F.; Overgaard, J. Nicotinamide pharmacokinetics in humans and mice: A comparative assessment and the implications for radiotherapy. Radiother. Oncol. 1993, 27, 131–139. [Google Scholar] [CrossRef]
- Hoffer, A. Biochemistry of nicotinic acid and nicotinamide. Psychosomatics 1967, 8, 95–100. [Google Scholar] [CrossRef]
- Greenbaum, C.J.; Kahn, S.E.; Palmer, J.P. Nicotinamide’s effects on glucose metabolism in subjects at risk for iddm. Diabetes 1996, 45, 1631–1634. [Google Scholar] [CrossRef]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of sirt1 in vitro, but can be a stimulator in cells. Cell. Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.L.; Boyer, J.L. Hepatic toxicity from large doses of vitamin b3 (nicotinamide). N. Engl. J. Med. 1973, 289, 1180–1182. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Bodeau, S.; Louvet, L.; Mary, A.; Boullier, A.; Lemaire-Hurtel, A.S.; Jonet, A.; Sonnet, P.; Kamel, S.; et al. N-methyl-2-pyridone-5-carboxamide (2py)-major metabolite of nicotinamide: An update on an old uremic toxin. Toxins (Basel) 2016, 8, 339. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Esper, N.E.; Brisset, S.; Mansour, J.; Lemaire-Hurtel, A.S.; Mary, A.; Brazier, M.; Kamel, S.; Mentaverri, R.; et al. Efficacy and safety of nicotinamide in haemodialysis patients: The nicoren study. Nephrol. Dial. Transplant. 2017, 32, 1597. [Google Scholar] [CrossRef]
- Jiao, X.; Doamekpor, S.K.; Bird, J.G.; Nickels, B.E.; Tong, L.; Hart, R.P.; Kiledjian, M. 5’ end nicotinamide adenine dinucleotide cap in human cells promotes rna decay through dxo-mediated denadding. Cell 2017, 168, 1015-1027.e10. [Google Scholar] [CrossRef]
- Zhou, S.S.; Li, D.; Zhou, Y.M.; Sun, W.P.; Liu, Q.G. B-vitamin consumption and the prevalence of diabetes and obesity among the us adults: Population based ecological study. BMC Public Health 2010, 10, 746. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of nad(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, M.; Schemainda, I. Fk866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Ghosh, D.; LeVault, K.R.; Barnett, A.J.; Brewer, G.J. A reversible early oxidized redox state that precedes macromolecular ros damage in aging nontransgenic and 3xtg-ad mouse neurons. J. Neurosci. 2012, 32, 5821–5832. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Jang, S.Y.; Kang, H.T.; Wei, B.; Jeoun, U.W.; Yoon, G.S.; Hwang, E.S. Modulation of mitochondrial membrane potential and ros generation by nicotinamide in a manner independent of sirt1 and mitophagy. Mol. Cells 2017, 40, 503–514. [Google Scholar]
- Aman, Y.; Qiu, Y.; Tao, J.; Fang, E.F. Therapeutic potential of boosting nad+ in aging and age-related diseases. Transl. Med. Aging 2018, 2, 30–37. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ros signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenet. 2016, 8, 61. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of foxo transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by sirt3-mediated sod2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3 Cell Metab-mediated deacetylation of evolutionarily conserved lysine 122 regulates mnsod activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mptp by sirt3-mediated deacetylation of cypd at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010, 2, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. Nicotinamide modulates mitochondrial membrane potential and cysteine protease activity during cerebral vascular endothelial cell injury. J. Vasc. Res. 2002, 39, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Chong, Z.Z.; Maiese, K. Nicotinamide: A nutritional supplement that provides protection against neuronal and vascular injury. J. Med. Food 2001, 4, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Lee, H.I.; Hwang, E.S. Nicotinamide extends replicative lifespan of human cells. Aging Cell 2006, 5, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high nad+/nadh ratio and sirt1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Hwang, E.S. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell 2009, 8, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Ok, J.S.; Song, S.B.; Hwang, E.S. Enhancement of replication and differentiation potential of human bone marrow stem cells by nicotinamide treatment. Int. J. Stem Cells 2018, 11, 13–25. [Google Scholar] [CrossRef]
- Berger, N.A. Poly(adp-ribose) in the cellular response to DNA damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef]
- Zhang, J.; Dawson, V.L.; Dawson, T.M.; Snyder, S.H. Nitric oxide activation of poly(adp-ribose) synthetase in neurotoxicity. Science 1994, 263, 687–689. [Google Scholar] [CrossRef]
- Ying, W.; Garnier, P.; Swanson, R.A. Nad+ repletion prevents parp-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem. Biophys. Res. Commun. 2003, 308, 809–813. [Google Scholar] [CrossRef]
- Zong, W.X.; Ditsworth, D.; Bauer, D.E.; Wang, Z.Q.; Thompson, C.B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004, 18, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, S.; Pioli, C. Multifaceted role of parp-1 in DNA repair and inflammation: Pathological and therapeutic implications in cancer and non-cancer diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, A. Treatment of arthritis by nicotinic acid and nicotinamide. Can. Med. Assoc. J. 1959, 81, 235–238. [Google Scholar] [PubMed]
- William, K. The Common form of Joint Dysfunction: Its Incidence and Treatment; E. L. Hildreth & Company: Brattleboro, VT, USA, 1949. [Google Scholar]
- Namazi, M.R. Nicotinamide: A potential addition to the anti-psoriatic weaponry. FASEB J. 2003, 17, 1377–1379. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.; Janjua, M.Z. The optimum dose of nicotinamide for protection of pancreatic beta-cells against the cytotoxic effect of streptozotocin in albino rat. J. Ayub Med. Coll. Abbottabad 2001, 13, 26–30. [Google Scholar]
- Kolb, H.; Burkart, V. Nicotinamide in type 1 diabetes. Mechanism of action revisited. Diabetes Care 1999, 22 (Suppl. 2), B16–B20. [Google Scholar]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of sirt1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of pgc-1alpha and sirt1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Ahn, J.; Cho, I.; Kim, S.; Kwon, D.; Ha, T. Dietary resveratrol alters lipid metabolism-related gene expression of mice on an atherogenic diet. J. Hepatol. 2008, 49, 1019–1028. [Google Scholar] [CrossRef]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.H.; Min, B.H.; Lee, K.M.; Cho, M.H.; Park, G.H.; Lee, K.H. Sirt1 promotes DNA repair activity and deacetylation of ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. Sirt1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of foxos and p53 by sirt1 modulators under oxidative stress. PLoS ONE 2013, 8, e73875. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.J.; Liu, N.; Xiao, Z.; Sun, T.; Wu, S.H.; Sun, W.X.; Xu, Z.G.; Yuan, H. Renal protective effect of sirtuin 1. J. Diabetes Res. 2014, 2014, 843786. [Google Scholar] [CrossRef]
- Delmez, J.A.; Slatopolsky, E. Hyperphosphatemia: Its consequences and treatment in patients with chronic renal disease. Am. J. Kidney Dis. 1992, 19, 303–317. [Google Scholar] [CrossRef]
- Thomas, P.; Dousa, S.A.K. Role of nicotinamide adenine dinucleotide (nad) in control of proximal renal tubular phosphate transport. In Urolithiasis; Springer: Boston, MA, USA, 1981; pp. 741–745. [Google Scholar]
- Eto, N.; Miyata, Y.; Ohno, H.; Yamashita, T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol. Dial. Transplant. 2005, 20, 1378–1384. [Google Scholar] [CrossRef]
- Rennie, G.; Chen, A.C.; Dhillon, H.; Vardy, J.; Damian, D.L. Nicotinamide and neurocognitive function. Nutr. Neurosci. 2015, 18, 193–200. [Google Scholar] [CrossRef]
- Slominski, A.; Semak, I.; Pisarchik, A.; Sweatman, T.; Szczesniewski, A.; Wortsman, J. Conversion of l-tryptophan to serotonin and melatonin in human melanoma cells. FEBS Lett. 2002, 511, 102–106. [Google Scholar] [CrossRef]
- McCarty, M.F. High-dose pyridoxine as an ’anti-stress’ strategy. Med. Hypotheses 2000, 54, 803–807. [Google Scholar] [CrossRef]
- Unilever. Niacinamide: Safety Assessment (Document Number d97/059), Section 5; Safety Assessment of Topically Applied Niacinamide; CTFA (Personal Care Products Council): Washington, DC, USA, 1998. [Google Scholar]
- Li, D.; Tian, Y.J.; Guo, J.; Sun, W.P.; Lun, Y.Z.; Guo, M.; Luo, N.; Cao, Y.; Cao, J.M.; Gong, X.J.; et al. Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats. Br. J. Nutr. 2013, 110, 2156–2164. [Google Scholar] [CrossRef]
- Griffin, S.M.; Pickard, M.R.; Orme, R.P.; Hawkins, C.P.; Fricker, R.A. Nicotinamide promotes neuronal differentiation of mouse embryonic stem cells in vitro. Neuroreport 2013, 24, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Pubchem. 13 Toxicity. National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nicotinamide (accessed on 23 April 2020).
- Lewis, C.M.; Canafax, D.M.; Sprafka, J.M.; Barbosa, J.J. Double-blind randomized trial of nicotinamide on early-onset diabetes. Diabetes Care 1992, 15, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Lopez, E.; Saleh-Gohari, N.; Helleday, T. Poly(adp-ribose) polymerase (parp-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003, 31, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Cefle, K.; Ucur, A.; Guney, N.; Ozturk, S.; Palanduz, S.; Tas, F.; Asoglu, O.; Bayrak, A.; Muslumanoglu, M.; Aydiner, A. Increased sister chromatid exchange frequency in young women with breast cancer and in their first-degree relatives. Cancer Genet. Cytogenet. 2006, 171, 65–67. [Google Scholar] [CrossRef]
- Utakoji, T.; Hosoda, K.; Umezawa, K.; Sawamura, M.; Matsushima, T.; Miwa, M.; Sugimura, T. Induction of sister chromatid exchanges by nicotinamide in chinese hamster lung fibroblasts and human lymphoblastoid cells. Biochem. Biophys. Res. Commun. 1979, 90, 1147–1152. [Google Scholar] [CrossRef]
- Oikawa, A.; Tohda, H.; Kanai, M.; Miwa, M.; Sugimura, T. Inhibitors of poly(adenosine diphosphate ribose) polymerase induce sister chromatid exchanges. Biochem. Biophys. Res. Commun. 1980, 97, 1311–1316. [Google Scholar] [CrossRef]
- Ishidate, M., Jr.; Harnois, M.C.; Sofuni, T. A comparative analysis of data on the clastogenicity of 951 chemical substances tested in mammalian cell cultures. Mutat. Res. 1988, 195, 151–213. [Google Scholar] [CrossRef]
- Lindahl-Kiessling, K.; Shall, S. Nicotinamide deficiency and benzamide-induced sister chromatid exchanges. Carcinogenesis 1987, 8, 1185–1188. [Google Scholar] [CrossRef]
- Ishidate, M., Jr.; Sofuni, T.; Yoshikawa, K.; Hayashi, M.; Nohmi, T.; Sawada, M.; Matsuoka, A. Primary mutagenicity screening of food additives currently used in japan. Food Chem. Toxicol. 1984, 22, 623–636. [Google Scholar] [CrossRef]
- Riklis, E.; Kol, R.; Marko, R. Trends and developments in radioprotection: The effect of nicotinamide on DNA repair. Int. J. Radiat. Biol. 1990, 57, 699–708. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, Y.; Li, L.; Wang, H.H.; Ma, X.S.; Qian, W.P.; Shen, W.; Schatten, H.; Sun, Q.Y. Sirt1, 2, 3 protect mouse oocytes from postovulatory aging. Aging (Albany NY) 2016, 8, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Rakieten, N.; Gordon, B.S.; Beaty, A.; Cooney, D.A.; Davis, R.D.; Schein, P.S. Pancreatic islet cell tumors produced by the combined action of streptozotocin and nicotinamide. Proc. Soc. Exp. Biol. Med. 1971, 137, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Schoental, R. The role of nicotinamide and of certain other modifying factors in diethylnitrosamine carcinogenesis: Fusaria mycotoxins and "spontaneous" tumors in animals and man. Cancer 1977, 40, 1833–1840. [Google Scholar] [CrossRef]
- Handler, P.; Dann, W.J. The inhibition of rat growth by nicotinamide. J. Biol. Chem. 1942, 146, 357–368. [Google Scholar]
- Handler, P. The effect of excessive nicotinamide feeding on rabbits and guinea pigs. J. Biol. Chem. 1944, 154, 203–206. [Google Scholar]
- Kang-Lee, Y.A.; McKee, R.W.; Wright, S.M.; Swendseid, M.E.; Jenden, D.J.; Jope, R.S. Metabolic effects of nicotinamide administration in rats. J. Nutr. 1983, 113, 215–221. [Google Scholar] [CrossRef]
- Mahmoud, Y.I.; Mahmoud, A.A. Role of nicotinamide (vitamin b3) in acetaminophen-induced changes in rat liver: Nicotinamide effect in acetaminophen-damged liver. Exp. Toxicol. Pathol. 2016, 68, 345–354. [Google Scholar] [CrossRef]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. Nnmt activation can contribute to the development of fatty liver disease by modulating the nad (+) metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef]
- Harrison, I.F.; Powell, N.M.; Dexter, D.T. The histone deacetylase inhibitor nicotinamide exacerbates neurodegeneration in the lactacystin rat model of parkinson’s disease. J. Neurochem. 2019, 148, 136–156. [Google Scholar] [CrossRef]
- Rakieten, N.; Gordon, B.S.; Beaty, A.; Cooney, D.A.; Schein, P.S.; Dixon, R.L. Modification of renal tumorigenic effect of streptozotocin by nicotinamide: Spontaneous reversibility of streptozotocin diabetes. Proc. Soc. Exp. Biol. Med. 1976, 151, 356–361. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, W.; Miner, J.; Fromm, M. Cross regulation of sirtuin 1, ampk, and ppargamma in conjugated linoleic acid treated adipocytes. PLoS ONE 2012, 7, e48874. [Google Scholar] [CrossRef]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/pbef/visfatin regulates insulin secretion in beta cells as a systemic nad biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Both, G.W.; Furuichi, Y.; Shatkin, A.J. 5’-terminal 7-methylguanosine in eukaryotic mrna is required for translation. Nature 1975, 255, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Rupprecht, K.M.; Hecht, S.M.; Shatkin, A.J. Eukaryotic mrna cap binding protein: Purification by affinity chromatography on sepharose-coupled m7gdp. Proc. Natl. Acad. Sci. USA 1979, 76, 4345–4349. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Douek, I.F.; Moore, W.P.; Gillmor, H.A.; McLean, A.E.; Bingley, P.J.; Gale, E.A.; European Nicotinamide Diabetes Intervention Trial Group. Safety of high-dose nicotinamide: A review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Felsted, R.L.; Chaykin, S. N1-methylnicotinamide oxidation in a number of mammals. J. Biol. Chem. 1967, 242, 1274–1279. [Google Scholar] [PubMed]
- Mrochek, J.E.; Jolley, R.L.; Young, D.S.; Turner, W.J. Metabolic response of humans to ingestion of nicotinic acid and nicotinamide. Clin. Chem. 1976, 22, 1821–1827. [Google Scholar] [CrossRef]
- Attwood, J.T.; Yung, R.L.; Richardson, B.C. DNA methylation and the regulation of gene transcription. Cell. Mol. Life Sci. 2002, 59, 241–257. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Drong, A.W.; Lindgren, C.M.; McCarthy, M.I. The genetic and epigenetic basis of type 2 diabetes and obesity. Clin. Pharmacol. Ther. 2012, 92, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Teyssier, C.; Strahl, B.D.; Stallcup, M.R. Role of protein methylation in regulation of transcription. Endocr. Rev. 2005, 26, 147–170. [Google Scholar] [CrossRef]
- Finkelstein, J.D.; Martin, J.J. Homocysteine. Int. J. Biochem. Cell Biol. 2000, 32, 385–389. [Google Scholar] [CrossRef]
- Cosmetic Ingredient Review Expert Panel. Final report of the safety assessment of niacinamide and niacin. Int. J. Toxicol. 2005, 24 (Suppl. 5), 1–31. [Google Scholar]
- Pissios, P. Nicotinamide n-methyltransferase: More than a vitamin b3 clearance enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.J.; Li, D.; Ma, Q.; Gu, X.Y.; Guo, M.; Lun, Y.Z.; Sun, W.P.; Wang, X.Y.; Cao, Y.; Zhou, S.S. Excess nicotinamide increases plasma serotonin and histamine levels. Sheng Li Xue Bao 2013, 65, 33–38. [Google Scholar] [PubMed]
- Burgos, E.S.; Schramm, V.L. Weak coupling of atp hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry 2008, 47, 11086–11096. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Tsuchiya, M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS ONE 2011, 6, e22781. [Google Scholar] [CrossRef]
- Zhou, S.S.; Li, D.; Sun, W.P.; Guo, M.; Lun, Y.Z.; Zhou, Y.M.; Xiao, F.C.; Jing, L.X.; Sun, S.X.; Zhang, L.B.; et al. Nicotinamide overload may play a role in the development of type 2 diabetes. World J. Gastroenterol. 2009, 15, 5674–5684. [Google Scholar] [CrossRef]
- Kannt, A.; Rajagopal, S.; Kadnur, S.V.; Suresh, J.; Bhamidipati, R.K.; Swaminathan, S.; Hallur, M.S.; Kristam, R.; Elvert, R.; Czech, J.; et al. A small molecule inhibitor of nicotinamide n-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 2018, 8, 3660. [Google Scholar] [CrossRef]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Riederer, M.; Erwa, W.; Zimmermann, R.; Frank, S.; Zechner, R. Adipose tissue as a source of nicotinamide n-methyltransferase and homocysteine. Atherosclerosis 2009, 204, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Hemati, T.; Moghadami-Tabrizi, N.; Davari-Tanha, F.; Salmanian, B.; Javadian, P. High plasma homocysteine and insulin resistance in patients with polycystic ovarian syndrome. Iran. J. Reprod. Med. 2011, 9, 223–228. [Google Scholar]
- Yang, N.; Yao, Z.; Miao, L.; Liu, J.; Gao, X.; Fan, H.; Hu, Y.; Zhang, H.; Xu, Y.; Qu, A.; et al. Novel clinical evidence of an association between homocysteine and insulin resistance in patients with hypothyroidism or subclinical hypothyroidism. PLoS ONE 2015, 10, e0125922. [Google Scholar] [CrossRef]
- Akintunde, A.; Nondi, J.; Gogo, K.; Jones, E.S.W.; Rayner, B.L.; Hackam, D.G.; Spence, J.D. Physiological phenotyping for personalized therapy of uncontrolled hypertension in africa. Am. J. Hypertens. 2017, 30, 923–930. [Google Scholar] [CrossRef]
- Feng, X.; Xu, Y. Hyperhomocysteinemia as a metabolic risk factor for glucose intolerance among high-risk groups of Chinese adults. Med. Sci. Monit. 2017, 23, 2775–2781. [Google Scholar] [CrossRef]
- Cardellini, M.; Perego, L.; D’Adamo, M.; Marini, M.A.; Procopio, C.; Hribal, M.L.; Andreozzi, F.; Frontoni, S.; Giacomelli, M.; Paganelli, M.; et al. C-174g polymorphism in the promoter of the interleukin-6 gene is associated with insulin resistance. Diabetes Care 2005, 28, 2007–2012. [Google Scholar] [CrossRef]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Mooney, R.A. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 2002, 51, 3391–3399. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Jiang, C.; Xu, M.; Pang, Y.; Feng, J.; Xiang, X.; Kong, W.; Xu, G.; Li, Y.; et al. Hyperhomocysteinemia promotes insulin resistance by inducing endoplasmic reticulum stress in adipose tissue. J. Biol. Chem. 2013, 288, 9583–9592. [Google Scholar] [CrossRef]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Asp. Med. 2019, 66, 49–61. [Google Scholar] [CrossRef]
- Xu, J.; Xu, S.Q.; Liang, J.; Lu, Y.; Luo, J.H.; Jin, J.H. Protective effect of nicotinamide in a mouse parkinson’s disease model. Zhejiang Da Xue Xue Bao Yi Xue Ban 2012, 41, 146–152. [Google Scholar] [PubMed]
- Williams, A.C.; Ramsden, D.B. Autotoxicity, methylation and a road to the prevention of parkinson’s disease. J. Clin. Neurosci. 2005, 12, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Aoyama, K.; Suno, M.; Awaya, T. N-methylation underlying parkinson’s disease. Neurotoxicol. Teratol. 2002, 24, 593–598. [Google Scholar] [CrossRef]
- Parsons, R.B.; Smith, S.W.; Waring, R.H.; Williams, A.C.; Ramsden, D.B. High expression of nicotinamide n-methyltransferase in patients with idiopathic parkinson’s disease. Neurosci. Lett. 2003, 342, 13–16. [Google Scholar] [CrossRef]
- Fukushima, T. Niacin metabolism and parkinson’s disease. Environ. Health Prev. Med. 2005, 10, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Kaetsu, A.; Lim, H.; Moriyama, M. Possible role of 1-methylnicotinamide in the pathogenesis of parkinson’s disease. Exp. Toxicol. Pathol. 2002, 53, 469–473. [Google Scholar] [CrossRef]
- Liu, M.; Chu, J.; Gu, Y.; Shi, H.; Zhang, R.; Wang, L.; Chen, J.; Shen, L.; Yu, P.; Chen, X.; et al. Serum n1-methylnicotinamide is associated with coronary artery disease in chinese patients. J. Am. Heart Assoc. 2017, 6, e004328. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Shai, I.; Stampfer, M.J.; Ma, J.; Manson, J.E.; Hankinson, S.E.; Cannuscio, C.; Selhub, J.; Curhan, G.; Rimm, E.B. Homocysteine as a risk factor for coronary heart diseases and its association with inflammatory biomarkers, lipids and dietary factors. Atherosclerosis 2004, 177, 375–381. [Google Scholar] [CrossRef]
- Zhang, R.; Shen, Y.; Zhou, L.; Sangwung, P.; Fujioka, H.; Zhang, L.; Liao, X. Short-term administration of nicotinamide mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell. Cardiol. 2017, 112, 64–73. [Google Scholar] [CrossRef]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide n-methyltransferase. Cdna cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [PubMed]
- Parsons, W.B., Jr. Studies of nicotinic acid use in hypercholesteremia. Changes in hepatic function, carbohydrate tolerance, and uric acid metabolism. Arch. Intern. Med. 1961, 107, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Volpi, E.; Lucidi, P.; Cruciani, G.; Monacchia, F.; Reboldi, G.; Brunetti, P.; Bolli, G.B.; De Feo, P. Nicotinamide counteracts alcohol-induced impairment of hepatic protein metabolism in humans. J. Nutr. 1997, 127, 2199–2204. [Google Scholar] [CrossRef]
- Strom, K.; Morales-Alamo, D.; Ottosson, F.; Edlund, A.; Hjort, L.; Jorgensen, S.W.; Almgren, P.; Zhou, Y.; Martin-Rincon, M.; Ekman, C.; et al. N(1)-methylnicotinamide is a signalling molecule produced in skeletal muscle coordinating energy metabolism. Sci. Rep. 2018, 8, 3016. [Google Scholar] [CrossRef]
- Kannt, A.; Pfenninger, A.; Teichert, L.; Tonjes, A.; Dietrich, A.; Schon, M.R.; Kloting, N.; Bluher, M. Association of nicotinamide-n-methyltransferase mrna expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance. Diabetologia 2015, 58, 799–808. [Google Scholar] [CrossRef]
- Hong, S.; Moreno-Navarrete, J.M.; Wei, X.; Kikukawa, Y.; Tzameli, I.; Prasad, D.; Lee, Y.; Asara, J.M.; Fernandez-Real, J.M.; Maratos-Flier, E.; et al. Nicotinamide n-methyltransferase regulates hepatic nutrient metabolism through sirt1 protein stabilization. Nat. Med. 2015, 21, 887–894. [Google Scholar] [CrossRef]
- Takeuchi, K.; Yokouchi, C.; Goto, H.; Umehara, K.; Yamada, H.; Ishii, Y. Alleviation of fatty liver in a rat model by enhancing n(1)-methylnicotinamide bioavailability through aldehyde oxidase inhibition. Biochem. Biophys. Res. Commun. 2018, 507, 203–210. [Google Scholar] [CrossRef]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-methylnicotinamide (mna), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef]
- Menon, R.M.; Adams, M.H.; Gonzalez, M.A.; Tolbert, D.S.; Leu, J.H.; Cefali, E.A. Plasma and urine pharmacokinetics of niacin and its metabolites from an extended-release niacin formulation. Int. J. Clin. Pharmacol. Ther. 2007, 45, 448–454. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-methyl-2-pyridone-5-carboxamide: A novel uremic toxin? Kidney Int. 2003, 63, S19–S21. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Martin, A.J.; Choy, B.; Fernandez-Penas, P.; Dalziell, R.A.; McKenzie, C.A.; Scolyer, R.A.; Dhillon, H.M.; Vardy, J.L.; Kricker, A.; et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N. Engl. J. Med. 2015, 373, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.; Silverberg, J.D.; Nguyen, T.T. Nicotinic acid-induced toxicity associated with cytopenia and decreased levels of thyroxine-binding globulin. Mayo Clin. Proc. 1992, 67, 465–468. [Google Scholar] [CrossRef]
- Tian, Y.J.; Luo, N.; Chen, N.N.; Lun, Y.Z.; Gu, X.Y.; Li, Z.; Ma, Q.; Zhou, S.S. Maternal nicotinamide supplementation causes global DNA hypomethylation, uracil hypo-incorporation and gene expression changes in fetal rats. Br. J. Nutr. 2014, 111, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, M.; Chan, X.Y.; Tan, S.Y.; Subramaniam, S.; Fan, Y.; Loh, E.; Chang, K.T.E.; Tan, T.C.; Chen, Q. Uncovering the mystery of opposite circadian rhythms between mouse and human leukocytes in humanized mice. Blood 2017, 130, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Otterness, D.M.; Weinshilboum, R.M. Human nicotinamide n-methyltransferase pharmacogenetics: Gene sequence analysis and promoter characterization. Pharmacogenetics 1999, 9, 307–316. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Effects | Examples of Effects | References |
|---|---|---|
| Protection against ATP depletion | [2] | |
| Decreased AD pathology and cognitive decline | [10] | |
| Improved sensory and motor neurological behavior | [3] | |
| Increased recovery from bilateral frontal brain injury | [4] | |
| Neuroprotection | Prevention/delay of ischemic stroke in stroke-prone hypertensive rats | [5] |
| Reduced lateral geniculate nucleus neuronal death | [6] | |
| Attenuated hippocampal neuronal death after global ischemia | [7] | |
| Improved motor deficits associated with Huntington’s disease phenotype | [8] | |
| Increased NAD+ level and mitochondrial function | [9] | |
| Amelioration of depression and psychological disorders | Amelioration of depression | [28] |
| Increased social interaction | [11] | |
| Anti-inflammation | Attenuated neutrophil recruitment in carrageenan-induced pleurisy or in lesions of autoimmune disease | [12,13] |
| Reduced arthritis activity | [14] | |
| Protection against vision and hearing loss | Attenuated retinal pigment cell death and age-related macular degeneration in animals | [29] |
| Reduced incidence of optic nerve degeneration and glaucoma | [30,31] | |
| Immune modulation | Improved mouse survival after lethal Staphylococcus enterotoxin B challenge | [32] |
| Skin protection/anti- skin disorders/cosmetic effects | Downregulation of the expression of inflammatory cytokines and protection against UV light | [33] |
| Anti-fibrosis | Attenuated development of pulmonary fibrosis | [34,35] |
| Anti-metastasis and adjuvant cancer therapy | Decreased growth and progression of bladder tumors | [36,37] |
| Photo-protection and reduced incidence of skin cancers | [15] | |
| Anti-HIV and -AIDS | Decreased provirus integration | [16] |
| Decreased viral RNA expression | [17] |
| Affected Organs and Conditions 2 | Observed Effects | Dose and Duration | References |
|---|---|---|---|
| Beneficial effects | |||
| Joints | Reduced itching in uremic patients | 550 mg twice a day (4 weeks) | [38] |
| pancreatic β-cell | β-cell function preserved and improved | 25 mg/kg daily intake (4 weeks) | [18,39] |
| Reduced the rate of diabetes incidence | 500 mg twice per day (2.5 years) | [19] | |
| No effect on the incidence of being diabetes-free | 1200 mg daily intake (5 years) | [20] | |
| Ineffective in prevention or delaying clinical onset of diabetes | 1.2 g daily intake (3 years) | [21] | |
| Skin | Reduced acne lesions and severity | 4% gel applied twice daily (8 weeks) | [26] |
| Attenuated immunosuppression with alterations in metabolism and apoptosis | 5% lotion applied before UV exposure | [40] | |
| Psychology | Improvements against depression | 0.5–1.5 g daily intake (3 weeks) | [22] |
| Relief from anxiety | A dose of 2 ug 3 h prior to test | [23] | |
| Kidney | Lowered serum concentrations of phosphorus, parathyroid hormone, and LDL, and increased serum HDL | 500 mg/day (with and increment every 2 weeks) (12 weeks) | [41] |
| Skin cancers non-melanoma | Reduced incidence of various types of skin cancers and actinic keratoses | 500 mg twice daily (4 months) | [42] |
| Adverse Effects | |||
| Minor effects | Frontal dull headaches, nausea, headache, dizziness | 1–18 g immediate | [43,44] |
| Pancreatic β-cell/plasma | Decreased insulin sensitivity, increased oxidative stress (H2O2) | 2 g daily (2 weeks) | [45,46] |
| Liver | Parenchymal-cell injury, portal fibrosis and cholestasis, liver injury | 3, 9 g daily (10 days) | [47] |
| Lymphocytes, platelets | Uremic toxicity-related cancer and thrombocytopenia | 1300, 1500 mg daily (24 weeks) | [48] |
| Kidney/platelets | Decreased serum phosphorus and thrombocytopenia | 0.52–2 g daily (3–6 months) | [49,50] |
| Subjects | Examples of Effects | Dose | Duration | Ref. |
|---|---|---|---|---|
| Death of mouse embryonic stem cells | 20 mM | 3–4 days | [95] | |
| Tumorigenicity. DNA damage, and sister chromatid exchanges | 1–10 mM 10 mM | 3 h 40 h | [100,101] | |
| Cells | 25 mM | 48 h | [102] | |
| Decreased SIRT1 activity. Increased intracellular ROS, spindle defects, and mitochondria dysfunction | 5 mM | 6, 12, 24 h | [106] | |
| Blocked mitochondria-related transcription. Worsened motor disturbance in Huntington’s disease model | 0.5, 1 mM | 96 h | [9] | |
| Mice and Rats | Oxidative DNA damage in hepatic and renal tissues. Impaired glucose tolerance and insulin sensitivity | 1 or 4 g/kg, d.w. | 8 weeks | [94] |
| Increased lethality | 4.5 g/kg, d.w., 2.5 g/kg, i.p. | 40 days | [44] | |
| Occurrence of pancreatic islet cell tumor | 350 mg/kg, i.p. | 226 days | [107] | |
| Increased incidence of kidney tumors | 350 mg/kg, i.p. | until die | [108] | |
| Decreased growth rate | 1, 2 %, d.w. 1, 2 %, d.w. | 24 days 20 days | [109,110] | |
| Growth inhibition, methyl deficiency, reduced tissue choline level, and increased hepatic lipids | 6, 20, 60 mg/100 g bw, i.p. | 2, 5 weeks | [111] | |
| Amelioration of acetaminophen-induced biochemical changes but occurrence of hepatotoxicity in healthy animals | 500 mg/kg, i.p. | 1.5 h | [112] | |
| Development of hepatic steatosis and fibrosis | 1%, d.w. | 6 weeks, 7 months | [113] | |
| Neurodegeneration of dopaminergic neurons Behavioral deficits and structural brain changes | 500 mg/kg, i.p. | 28 days | [114] | |
| Blocked mitochondrial-related transcription, worsened motor phenotype | 250mg/kg/day, s.c. | 28 days | [9] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, E.S.; Song, S.B. Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules 2020, 10, 687. https://doi.org/10.3390/biom10050687
Hwang ES, Song SB. Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules. 2020; 10(5):687. https://doi.org/10.3390/biom10050687
Chicago/Turabian StyleHwang, Eun Seong, and Seon Beom Song. 2020. "Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment" Biomolecules 10, no. 5: 687. https://doi.org/10.3390/biom10050687
APA StyleHwang, E. S., & Song, S. B. (2020). Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules, 10(5), 687. https://doi.org/10.3390/biom10050687