Targeting Intrinsically Disordered Proteins through Dynamic Interactions


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Characterization of Disordered Protein Ensembles: A Crucial Role for Atomistic Simulations
2.1. Fundamental Challenges in Experimental Determination of Disordered Protein Ensembles
2.2. Recent Advances in de Novo Simulations of Disordered Protein Ensembles
2.2.1. Overcoming Sampling Bottleneck using Enhanced Sampling and GPU Computing
2.2.2. Balanced Explicit Protein Force Fields for Describing Disordered Protein Ensembles
2.2.3. Implicit Solvent as a Promising Alternative for Simulating Disordered Ensembles
3. Modulating Disordered Protein Ensembles via Dynamic Interactions
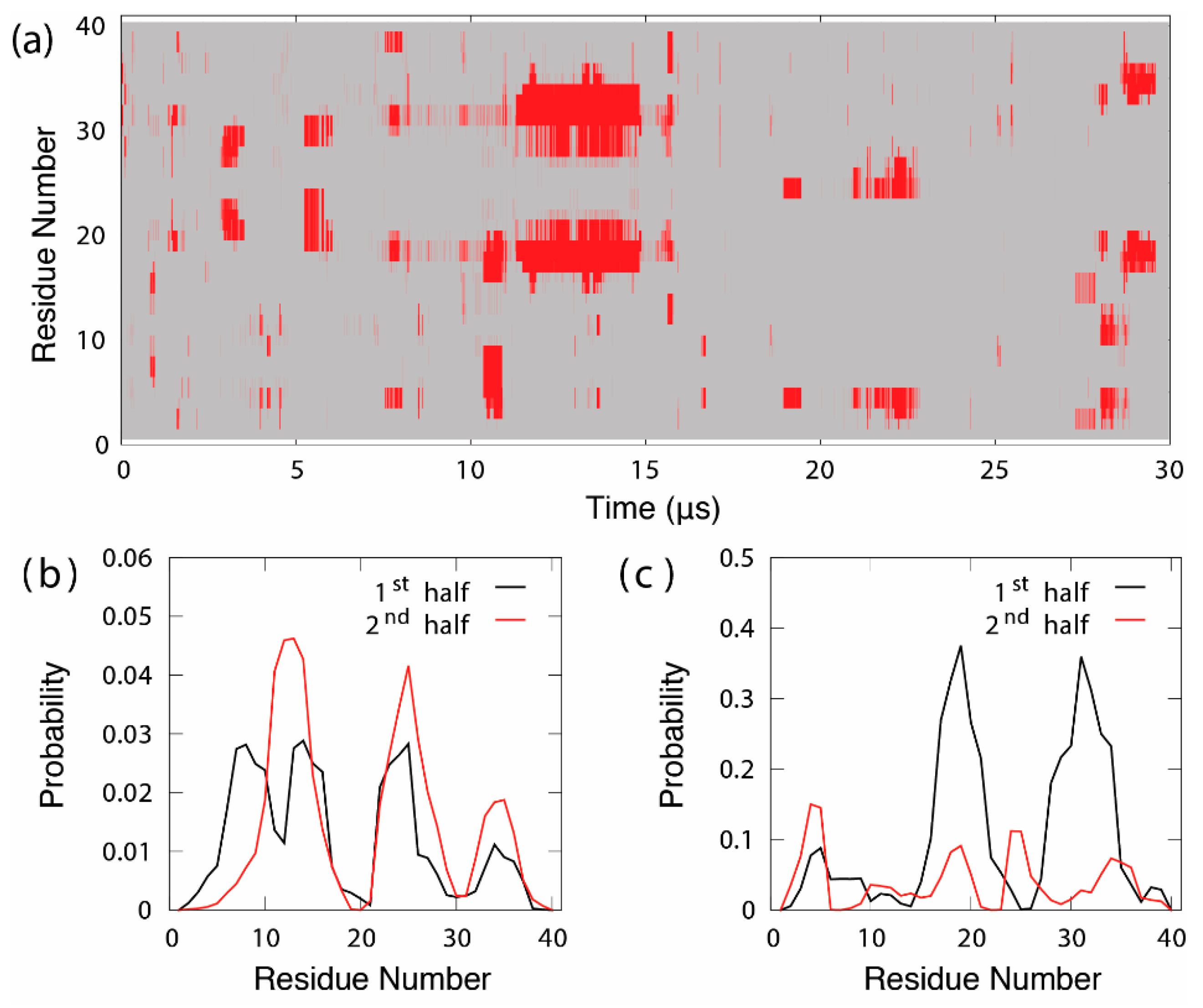
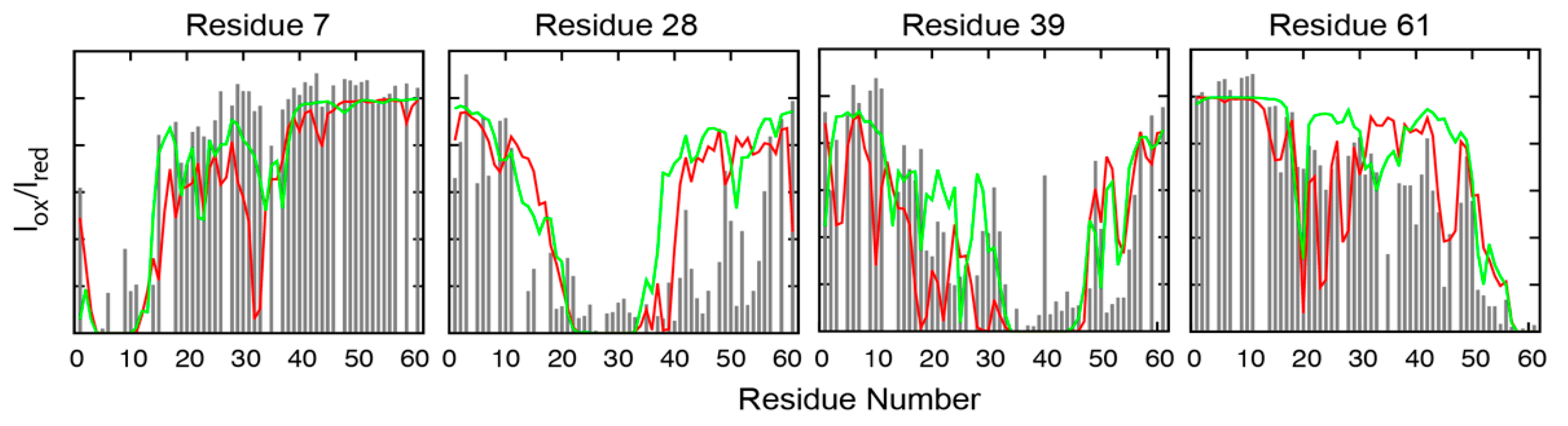
3.1. Dynamic Interactions of c-Myc Inhibitors
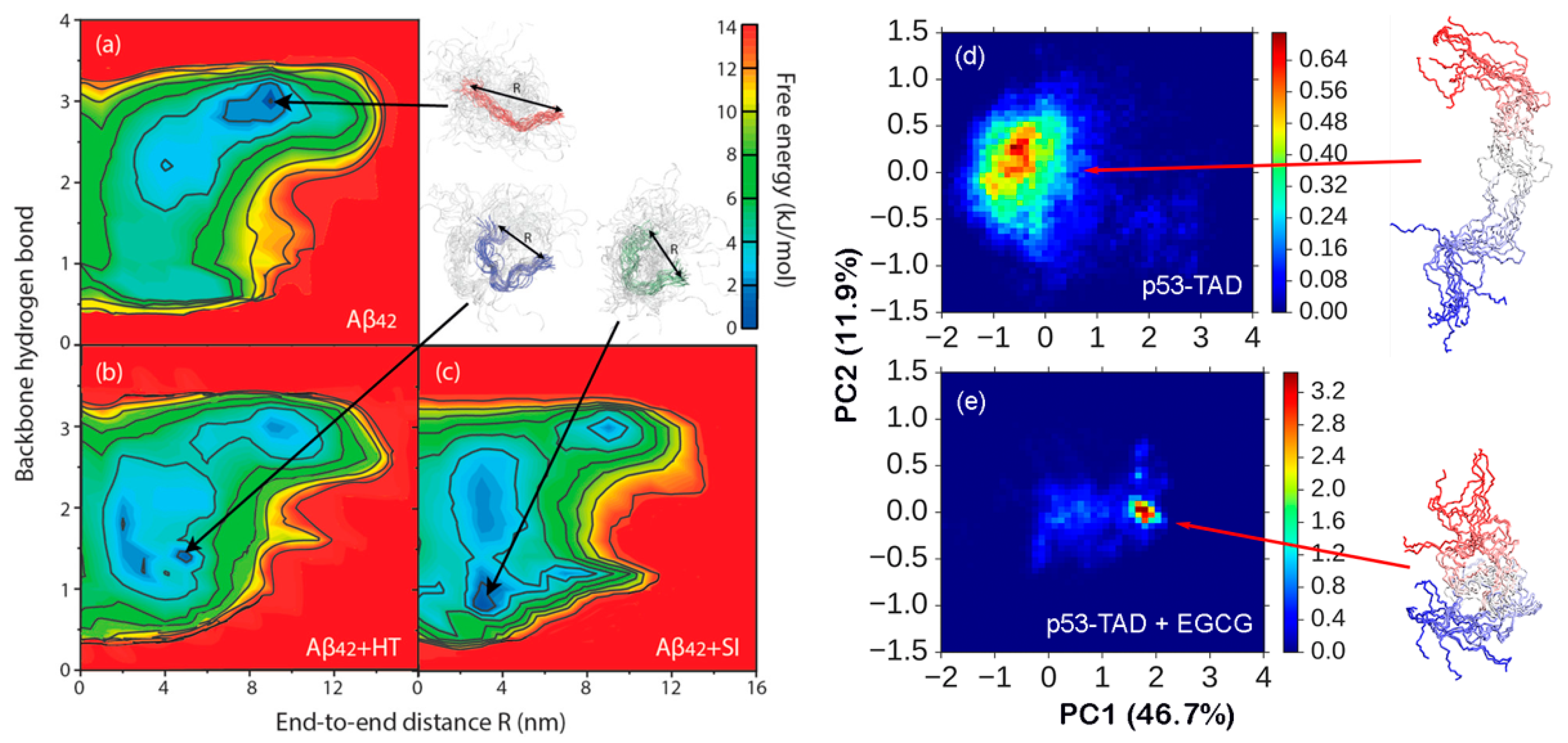
3.2. Inhibition of Aggregation by Induced Compaction
3.3. Modulating Regulatory IDPs via Dynamic Interactions
4. Concluding Discussions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets—The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Romero, P.; Obradovic, Z.; Li, X.H.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins-Struct. Funct. Genet. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Smock, R.G.; Gierasch, L.M. Sending signals dynamically. Science 2009, 324, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Hilser, V.J.; Thompson, E.B. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 8311–8315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.T.; Li, J.; Grasso, E.; Wrabl, J.O.; Hilser, V.J. Ensemble allosteric model: Energetic frustration within the intrinsically disordered glucocorticoid receptor. Philos. Trans. R. Soc. B-Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Li, M.D.; Liu, Z.R. A comprehensive ensemble model for comparing the allosteric effect of ordered and disordered proteins. PLoS Comput. Biol. 2018, 14, e1006393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J. Towards the physical basis of how intrinsic disorder mediates protein function. Arch. Biochem. Biophys. 2012, 524, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D-2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Tsafou, K.; Tiwari, P.B.; Forman-Kay, J.D.; Metallo, S.J.; Toretsky, J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018, 430, 2321–2341. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Kumar, D.; Sharma, N.; Uversky, V.N. Intrinsically Disordered Side of the Zika Virus Proteome. Front. Cell. Infect. Microbiol. 2016, 6, 144. [Google Scholar] [CrossRef]
- Hu, G.; Wu, Z.H.; Wang, K.; Uversky, V.N.; Kurgan, L. Untapped Potential of Disordered Proteins in Current Druggable Human Proteome. Curr. Drug Targets 2016, 17, 1198–1205. [Google Scholar] [CrossRef]
- Sluchanko, N.N.; Bustos, D.M. Intrinsic disorder associated with 14-3-3 proteins and their partners. Prog. Mol. Biol. Trans. Sci. 2019, 166, 19–61. [Google Scholar]
- Uversky, V.N. Intrinsic disorder here, there, and everywhere, and nowhere to escape from it. Cell. Mol. Life Sci. 2017, 74, 3065–3067. [Google Scholar] [CrossRef]
- Vacic, V.; Iakoucheva, L.M. Disease mutations in disordered regions--exception to the rule? Mol. BioSyst. 2012, 8, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittag, T.; Forman-Kay, J.D. Atomic-level characterization of disordered protein ensembles. Curr. Opin. Struct. Biol. 2007, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.K.; Stultz, C.M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 21, 426–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Click, T.H.; Ganguly, D.; Chen, J. Intrinsically Disordered Proteins in a Physics-Based World. Int. J. Mol. Sci. 2010, 11, 5292–5309. [Google Scholar] [CrossRef] [Green Version]
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Molecular recognition features (MoRFs) in three domains of life. Mol. BioSyst. 2016, 12, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef]
- Mustata, G.; Follis, A.V.; Hammoudeh, D.I.; Metallo, S.J.; Wang, H.; Prochownik, E.V.; Lazo, J.S.; Bahar, I. Discovery of novel Myc-Max heterodimer disruptors with a three-dimensional pharmacophore model. J. Med. Chem. 2009, 52, 1247–1250. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Iconaru, L.I.; Ban, D.; Bharatham, K.; Ramanathan, A.; Zhang, W.X.; Shelat, A.A.; Zuo, J.; Kriwacki, R.W. Discovery of Small Molecules that Inhibit the Disordered Protein, p27(Kip1). Sci Rep-Uk 2015, 5, 15686. [Google Scholar] [CrossRef] [Green Version]
- Ban, D.; Iconaru, L.I.; Ramanathan, A.; Zuo, J.; Kriwacki, R.W. A Small Molecule Causes a Population Shift in the Conformational Landscape of an Intrinsically Disordered Protein. J. Am. Chem. Soc. 2017, 139, 13692–13700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Cao, H.Q.; Liu, Z.R. Binding cavities and druggability of intrinsically disordered proteins. Protein Sci. 2015, 24, 688–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Han, K.H. PreSMo Target-Binding Signatures in Intrinsically Disordered Proteins. Mol. Cells 2018, 41, 889–899. [Google Scholar] [PubMed]
- Zhu, M.; De Simone, A.; Schenk, D.; Toth, G.; Dobson, C.M.; Vendruscolo, M. Identification of small-molecule binding pockets in the soluble monomeric form of the A beta 42 peptide. J. Chem. Phys. 2013, 139, 07B609_1. [Google Scholar] [CrossRef] [Green Version]
- Pickhardt, M.; Neumann, T.; Schwizer, D.; Callaway, K.; Vendruscolo, M.; Schenk, D.; St George-Hyslop, P.; Mandelkow, E.M.; Dobson, C.M.; McConlogue, L.; et al. Identification of Small Molecule Inhibitors of Tau Aggregation by Targeting Monomeric Tau As a Potential Therapeutic Approach for Tauopathies. Curr. Alzheimer Res. 2015, 12, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Chia, S.; Habchi, J.; Knowles, T.P.J.; Dobson, C.M.; Vendruscolo, M. A Fragment-Based Method of Creating Small-Molecule Libraries to Target the Aggregation of Intrinsically Disordered Proteins. Acs Comb. Sci. 2016, 18, 144–153. [Google Scholar] [CrossRef]
- Yu, C.; Niu, X.G.; Jin, F.; Liu, Z.R.; Jin, C.W.; Lai, L.H. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Bayliss, R.; Burgess, S.G.; Leen, E.; Richards, M.W. A moving target: Structure and disorder in pursuit of Myc inhibitors. Biochem. Soc. Trans. 2017, 45, 709–717. [Google Scholar] [CrossRef]
- Chong, B.; Li, M.D.; Li, T.; Yu, M.; Zhang, Y.G.; Liu, Z.R. Conservation of Potentially Druggable Cavities in Intrinsically Disordered Proteins. Acs Omega 2018, 3, 15643–15652. [Google Scholar] [CrossRef]
- De Mol, E.; Fenwick, R.B.; Phang, C.T.W.; Buzon, V.; Szulc, E.; de la Fuente, A.; Escobedo, A.; Garcia, J.; Bertoncini, C.W.; Estebanez-Perpina, E.; et al. EPI-001, A Compound Active against Castration-Resistant Prostate Cancer, Targets Transactivation Unit 5 of the Androgen Receptor. ACS Chem. Biol. 2016, 11, 2499–2505. [Google Scholar] [CrossRef]
- Toth, G.; Gardai, S.J.; Zago, W.; Bertoncini, C.W.; Cremades, N.; Roy, S.L.; Tambe, M.A.; Rochet, J.C.; Galvagnion, C.; Skibinski, G.; et al. Targeting the Intrinsically Disordered Structural Ensemble of alpha-Synuclein by Small Molecules as a Potential Therapeutic Strategy for Parkinson’s Disease. PLoS ONE 2014, 9, e8713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygren, P.J.; Mehta, S.; Schweppe, D.K.; Langeberg, L.K.; Whiting, J.L.; Weisbrod, C.R.; Bruce, J.E.; Zhang, J.; Veesler, D.; Scott, J.D. Intrinsic disorder within AKAP79 fine-tunes anchored phosphatase activity toward substrates and drug sensitivity. Elife 2017, 6. [Google Scholar] [CrossRef]
- Basu, S.; Soderquist, F.; Wallner, B. Proteus: A random forest classifier to predict disorder-to-order transitioning binding regions in intrinsically disordered proteins. J. Comput.-Aided Mol. Des. 2017, 31, 453–466. [Google Scholar] [CrossRef]
- Heller, G.T.; Bonomi, M.; Vendruscolo, M. Structural Ensemble Modulation upon Small-Molecule Binding to Disordered Proteins. J. Mol. Biol. 2018, 430, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Sun, Q.; Zhang, W.L.; Liu, Y.; Lai, L.H. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Blackledge, M.; Zweckstetter, M. Intrinsically Disordered Proteins: From Sequence and Conformational Properties toward Drug Discovery. ChemBioChem 2012, 13, 930–950. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Uversky, V.N. Drugs for ’protein clouds’: Targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 2010, 10, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin. Drug Discov. 2012, 7, 475–488. [Google Scholar] [CrossRef]
- Chen, C.Y.C.; Tou, W.L. How to design a drug for the disordered proteins. Drug Discov. Today 2013, 18, 910–915. [Google Scholar] [CrossRef]
- Heller, G.T.; Sormanni, P.; Vendruscolo, M. Targeting disordered proteins with small molecules using entropy. Trends Biochem. Sci. 2015, 40, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, P.; Vendruscolo, M. Druggability of Intrinsically Disordered Proteins. In Intrinsically Disordered Proteins Studied by Nmr Spectroscopy; Felli, I.C., Pierattelli, R., Eds.; Springer: Cham, Switzerland, 2015; Volume 870, pp. 383–400. [Google Scholar]
- Sammak, S.; Zinzalla, G. Targeting protein-protein interactions (PPIs) of transcription factors: Challenges of intrinsically disordered proteins (IDPs) and regions (IDRs). Prog. Biophys. Mol. Biol. 2015, 119, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2016, 11, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K.; Lo, S.C.; Wang, Y.S.; Hou, M.H. Recent insights into the development of therapeutics against coronavirus diseases by targeting N protein. Drug Discov. Today 2016, 21, 562–572. [Google Scholar] [CrossRef]
- Varadi, M.; Kosol, S.; Lebrun, P.; Valentini, E.; Blackledge, M.; Dunker, A.K.; Felli, I.C.; Forman-Kay, J.D.; Kriwacki, R.W.; Pierattelli, R.; et al. pE-DB: A database of structural ensembles of intrinsically disordered and of unfolded proteins. Nucleic Acids Res. 2014, 42, D326–D335. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Felli, I.C.; Pierattelli, R. Intrinsically Disordered Proteins Studied by NMR Spectroscopy, 1st ed.; Springer International Publishing: Cham, Switzerland, 2015; Volume 870. [Google Scholar]
- Ganguly, D.; Chen, J. Structural interpretation of paramagnetic relaxation enhancement-derived distances for disordered protein states. J. Mol. Biol. 2009, 390, 467–477. [Google Scholar] [CrossRef]
- Bonomi, M.; Heller, G.T.; Camilloni, C.; Vendruscolo, M. Principles of protein structural ensemble determination. Curr. Opin. Struct. Biol. 2017, 42, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Ravera, E.; Sgheri, L.; Parigi, G.; Luchinat, C. A critical assessment of methods to recover information from averaged data. Phys. Chem. Chem. Phys. 2016, 18, 5686–5701. [Google Scholar] [CrossRef]
- Marsh, J.A.; Neale, C.; Jack, F.E.; Choy, W.Y.; Lee, A.Y.; Crowhurst, K.A.; Forman-Kay, J.D. Improved structural characterizations of the drkN SH3 domain unfolded state suggest a compact ensemble with native-like and non-native structure. J. Mol. Biol. 2007, 367, 1494–1510. [Google Scholar] [CrossRef]
- Salmon, L.; Nodet, G.; Ozenne, V.; Yin, G.W.; Jensen, M.R.; Zweckstetter, M.; Blackledge, M. NMR Characterization of Long-Range Order in Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2010, 132, 8407–8418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, K.; Castaneda, C.A.; Schneidman-Duhovny, D.; Sali, A.; Nava-Tudela, A.; Fushman, D. Recovering a Representative Conformational Ensemble from Underdetermined Macromolecular Structural Data. J. Am. Chem. Soc. 2013, 135, 16595–16609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, S.; Shin, J.H.; Raghunandan, S.; Curtis, J.E.; Kelman, Z. Atomistic Ensemble Modeling and Small-Angle Neutron Scattering of Intrinsically Disordered Protein Complexes: Applied to Minichromosome Maintenance Protein. Biophys. J. 2011, 101, 2999–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, S.J.; Wright, D.W.; Zhang, H.L.; Brookes, E.H.; Chen, J.H.; Irving, T.C.; Krueger, S.; Barlow, D.J.; Edler, K.J.; Scott, D.J.; et al. Atomistic modelling of scattering data in the Collaborative Computational Project for Small Angle Scattering (CCP-SAS). J. Appl. Crystallogr. 2016, 49, 1861–1875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.H.; Howell, S.C.; Wright, D.W.; Heindel, A.; Qiu, X.Y.; Chen, J.H.; Curtis, J.E. Combined Monte Carlo/torsion-angle molecular dynamics for ensemble modeling of proteins, nucleic acids and carbohydrates. J. Mol. Graphics Modell. 2017, 73, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kragelj, J.; Ozenne, V.; Blackledge, M.; Jensen, M.R. Conformational propensities of intrinsically disordered proteins from NMR chemical shifts. ChemPhysChem 2013, 14, 3034–3045. [Google Scholar] [CrossRef]
- Fisher, C.K.; Huang, A.; Stultz, C.M. Modeling intrinsically disordered proteins with bayesian statistics. J. Am. Chem. Soc. 2010, 132, 14919–14927. [Google Scholar] [CrossRef]
- Bertini, I.; Giachetti, A.; Luchinat, C.; Parigi, G.; Petoukhov, M.V.; Pierattelli, R.; Ravera, E.; Svergun, D.I. Conformational space of flexible biological macromolecules from average data. J. Am. Chem. Soc. 2010, 132, 13553–13558. [Google Scholar] [CrossRef]
- Levine, Z.A.; Shea, J.E. Simulations of disordered proteins and systems with conformational heterogeneity. Curr. Opin. Struct. Biol. 2017, 43, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Knott, M.; Best, R.B. A preformed binding interface in the unbound ensemble of an intrinsically disordered protein: Evidence from molecular simulations. PLoS Comput. Biol. 2012, 8, e1002605. [Google Scholar] [CrossRef]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, D.; Chen, J. Atomistic details of the disordered states of KID and pKID. implications in coupled binding and folding. J. Am. Chem. Soc. 2009, 131, 5214–5223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ganguly, D.; Chen, J. Residual structures, conformational fluctuations, and electrostatic interactions in the synergistic folding of two intrinsically disordered proteins. PLoS Comput. Biol. 2012, 8, e1002353. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Case, D.A.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; et al. Kollman AMBER 2017; University of California: San Francisco, CA, USA, 2017. [Google Scholar]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.-P.; Shukla, D.; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2012, 9, 461–469. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kal, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.H.; Chen, J.H. Accelerate Sampling in Atomistic Energy Landscapes Using Topology-Based Coarse-Grained Models. J. Chem. Theory Comput. 2014, 10, 918–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritsugu, K.; Terada, T.; Kidera, A. Scalable free energy calculation of proteins via multiscale essential sampling. J. Chem. Phys. 2010, 133, 12B603. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, A.; Lyle, N.; Harmon, T.S.; Pappu, R.V. Hamiltonian Switch Metropolis Monte Carlo Simulations for Improved Conformational Sampling of Intrinsically Disordered Regions Tethered to Ordered Domains of Proteins. J. Chem. Theory Comput. 2014, 10, 3550–3562. [Google Scholar] [CrossRef] [PubMed]
- Peter, E.K.; Shea, J.E. A hybrid MD-kMC algorithm for folding proteins in explicit solvent. Phys. Chem. Chem. Phys. 2014, 16, 6430–6440. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, J. Enhanced sampling and applications in protein folding in explicit solvent. J. Chem. Phys. 2010, 132, 244101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.Q.; Yang, W. Practically Efficient and Robust Free Energy Calculations: Double-Integration Orthogonal Space Tempering. J. Chem. Theory Comput. 2012, 8, 810–823. [Google Scholar] [CrossRef]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica exchange with solute scaling: A more efficient version of replica exchange with solute tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef] [Green Version]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Chen, J.H. Optimization of the GBMV2 implicit solvent force field for accurate simulation of protein conformational equilibria. J. Comput. Chem. 2017, 38, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Nerenberg, P.S.; Jo, B.; So, C.; Tripathy, A.; Head-Gordon, T. Optimizing Solute-Water van der Waals Interactions To Reproduce Solvation Free Energies. J. Phys. Chem. B 2012, 116, 4524–4534. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.M.; Pappu, R.V. Improvements to the ABSINTH Force Field for Proteins Based on Experimentally Derived Amino Acid Specific Backbone Conformational Statistics. J. Chem. Theory Comput. 2019, 15, 1367–1382. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J. Residual Structures and Transient Long-Range Interactions of p53 Transactivation Domain: Assessment of Explicit Solvent Protein Force Fields. J. Chem. Theory Comput. 2019, 15, 4708–4720. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Nerenberg, P.S.; Head-Gordon, T. Optimizing Protein-Solvent Force Fields to Reproduce Intrinsic Conformational Preferences of Model Peptides. J. Chem Theory. Comput. 2011, 7, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zheng, W.; Mittal, J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113–5124. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Li, D.-W.; Brüschweiler, R. Balanced Amino-Acid-Specific Molecular Dynamics Force Field for the Realistic Simulation of Both Folded and Disordered Proteins. J. Chem. Theory Comput. 2020, 16, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Best, R.B. Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154. [Google Scholar] [CrossRef]
- Ganguly, D.; Chen, J. Modulation of the disordered conformational ensembles of the p53 transactivation domain by cancer-associated mutations. PLoS Comput. Biol. 2015, 11, e1004247. [Google Scholar] [CrossRef] [Green Version]
- Pall, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. Lect. Notes Comput. Sci. 2015, 8759, 3–27. [Google Scholar]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Terakawa, T.; Kameda, T.; Takada, S. On Easy Implementation of a Variant of the Replica Exchange with Solute Tempering in GROMACS. J. Comput. Chem. 2011, 32, 1228–1234. [Google Scholar] [CrossRef]
- Bussi, G. Hamiltonian replica exchange in GROMACS: A flexible implementation. Mol. Phys. 2014, 112, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Im, W.; Brooks, C.L. Balancing solvation and intramolecular interactions: Toward a consistent generalized born force field. J. Am. Chem. Soc. 2006, 128, 3728–3736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Chiricotto, M.; Liu, X.; Nordquist, E.; Feig, M.; Brooks, C.L., 3rd; Chen, J. Accelerating the Generalized Born with Molecular Volume and Solvent Accessible Surface Area Implicit Solvent Model Using Graphics Processing Units. J. Comput. Chem. 2020, 41, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, S.; Birol, M.; Rhoades, E.; Miranker, A.D.; Levine, Z.A. Targeting the Intrinsically Disordered Proteome Using Small-Molecule Ligands. In Intrinsically Disordered Proteins; Rhoades, E., Ed.; Elsevier Inc.: London, UK, 2018; Volume 611, pp. 703–734. [Google Scholar]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- McDowell, C.; Chen, J.; Chen, J. Potential Conformational Heterogeneity of p53 Bound to S100B(betabeta). J. Mol. Biol. 2013, 425, 999–1010. [Google Scholar] [CrossRef] [Green Version]
- Weng, J.; Wang, W. Dynamic multivalent interactions of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2019, 62, 9–13. [Google Scholar] [CrossRef]
- Michel, J.; Cuchillo, R. The impact of small molecule binding on the energy landscape of the intrinsically disordered protein C-myc. PLoS ONE 2012, 7, e41070. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hammoudeh, D.I.; Follis, A.V.; Reese, B.E.; Lazo, J.S.; Metallo, S.J.; Prochownik, E.V. Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 2007, 6, 2399–2408. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Yu, C.; Lai, L.; Liu, Z. Ligand clouds around protein clouds: A scenario of ligand binding with intrinsically disordered proteins. PLoS Comput. Biol. 2013, 9, e1003249. [Google Scholar] [CrossRef] [Green Version]
- Heller, G.T.; Aprile, F.A.; Bonomi, M.; Camilloni, C.; De Simone, A.; Vendruscolo, M. Sequence Specificity in the Entropy-Driven Binding of a Small Molecule and a Disordered Peptide. J. Mol. Biol. 2017, 429, 2772–2779. [Google Scholar] [CrossRef]
- Daniels, M.J.; Nourse, J.B.; Kim, H.; Sainati, V.; Schiavina, M.; Murrali, M.G.; Pan, B.Y.; Ferrie, J.J.; Haney, C.M.; Moons, R.; et al. Cyclized NDGA modifies dynamic alpha-synuclein monomers preventing aggregation and toxicity. Sci. Rep. 2019, 9, 1–17. [Google Scholar]
- Liang, C.; Savinov, S.N.; Fejzo, J.; Eyles, S.J.; Chen, J. Modulation of Amyloid-beta42 Conformation by Small Molecules Through Nonspecific Binding. J. Chem. Theory Comput. 2019, 15, 5169–5174. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J. Modulation of p53 Transactivation Domain Conformations by Ligand Binding and Cancer-Associated Mutations. Pac. Symp. Biocomput. 2020, 25, 195–206. [Google Scholar]
- Du, G.J.; Zhang, Z.; Wen, X.D.; Yu, C.; Calway, T.; Yuan, C.S.; Wang, C.Z. Epigallocatechin Gallate (EGCG) is the most effective cancer chemopreventive polyphenol in green tea. Nutrients 2012, 4, 1679–1691. [Google Scholar] [CrossRef]
- Fujiki, H.; Sueoka, E.; Watanabe, T.; Suganuma, M. Primary cancer prevention by green tea, and tertiary cancer prevention by the combination of green tea catechins and anticancer compounds. J. Cancer Prev. 2015, 20, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.M.; Lee, D.H.; Seo, A.Y.; Lee, H.J.; Kim, S.B.; Son, W.C.; Kim, Y.K.; Lee, S.J.; Park, S.H.; Kim, N.; et al. Green tea extracts for the prevention of metachronous colorectal polyps among patients who underwent endoscopic removal of colorectal adenomas: A randomized clinical trial. Clin. Nutr. 2018, 37, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green tea epigallocatechin-3-gallate (EGCG) reduces beta-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 2008, 1214, 177–187. [Google Scholar] [CrossRef]
- Santofimia-Castano, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velazquez-Campoy, A.; Iovanna, J.L.; Neira, J.L. Designing and repurposing drugs to target intrinsically disordered proteins for cancer treatment: Using NUPR1 as a paradigm. Mol. Cell. Oncol. 2019, 6, e1612678. [Google Scholar] [CrossRef]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velazquez-Campoy, A.; Iovanna, J.L.; Abian, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holehouse, A.S.; Pappu, R.V. Functional Implications of Intracellular Phase Transitions. Biochemistry 2018, 57, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Heller, G.T.; Aprile, F.A.; Vendruscolo, M. Methods of probing the interactions between small molecules and disordered proteins. Cell. Mol. Life Sci. 2017, 74, 3225–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrev, V.S.; Fred, L.M.; Gerhart, K.P.; Metallo, S.J. Characterization of the Binding of Small Molecules to Intrinsically Disordered Proteins. In Intrinsically Disordered Proteins; Rhoades, E., Ed.; Elsevier Inc.: London, UK, 2018; Volume 611, pp. 677–702. [Google Scholar]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Liu, X.; Chen, J. Targeting Intrinsically Disordered Proteins through Dynamic Interactions. Biomolecules 2020, 10, 743. https://doi.org/10.3390/biom10050743
Chen J, Liu X, Chen J. Targeting Intrinsically Disordered Proteins through Dynamic Interactions. Biomolecules. 2020; 10(5):743. https://doi.org/10.3390/biom10050743
Chicago/Turabian StyleChen, Jianlin, Xiaorong Liu, and Jianhan Chen. 2020. "Targeting Intrinsically Disordered Proteins through Dynamic Interactions" Biomolecules 10, no. 5: 743. https://doi.org/10.3390/biom10050743
APA StyleChen, J., Liu, X., & Chen, J. (2020). Targeting Intrinsically Disordered Proteins through Dynamic Interactions. Biomolecules, 10(5), 743. https://doi.org/10.3390/biom10050743