Analysis of Protein Disorder Predictions in the Light of a Protein Structural Alphabet


Abstract
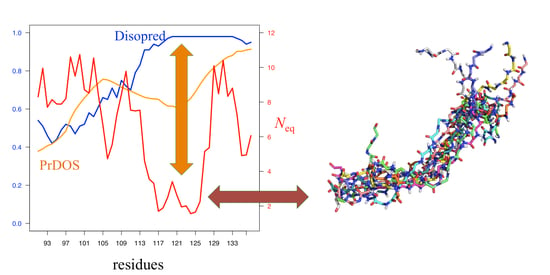
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Neq Entropy Index
2.3. Disorder Prediction from the Sequence
2.4. Analyses
3. Results
3.1. Data Analyses
3.2. Comparison of Neq with Prediction Results
3.3. General Tendencies
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eisenberg, D. The discovery of the alpha-helix and beta-sheet, the principal structural features of proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 11207–11210. [Google Scholar] [CrossRef] [Green Version]
- Unger, R.; Harel, D.; Wherland, S.; Sussman, J.L. A 3D building blocks approach to analyzing and predicting structure of proteins. Proteins 1989, 5, 355–373. [Google Scholar] [CrossRef]
- Offmann, B.; Tyagi, M.; de Brevern, A.G. Local Protein Structures. Curr. Bioinform. 2007, 3, 165–202. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhao, Y.P.; Zheng, W.M. CLEMAPS: Multiple alignment of protein structures based on conformational letters. Proteins 2008, 71, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Tung, C.H.; Huang, J.W.; Yang, J.M. Kappa-alpha plot derived structural alphabet and BLOSUM-like substitution matrix for rapid search of protein structure database. Genome Biol. 2007, 8, R31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, S.; Joseph, A.P.; Srinivasan, N.; Gelly, J.C.; de Brevern, A.G. mulPBA: An efficient multiple protein structure alignment method based on a structural alphabet. J. Biomol. Struct. Dyn. 2014, 32, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Pandini, A.; Fornili, A.; Fraternali, F.; Kleinjung, J. GSATools: Analysis of allosteric communication and functional local motions using a structural alphabet. Bioinformatics 2013, 29, 2053–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudev, M.; Lim, C. Discovering structural motifs using a structural alphabet: Application to magnesium-binding sites. BMC Bioinform. 2007, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Narwani, T.J.; Etchebest, C.; Craveur, P.; Leonard, S.; Rebehmed, J.; Srinivasan, N.; Bornot, A.; Gelly, J.C.; de Brevern, A.G. In silico prediction of protein flexibility with local structure approach. Biochimie 2019, 165, 150–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karchin, R.; Cline, M.; Mandel-Gutfreund, Y.; Karplus, K. Hidden Markov models that use predicted local structure for fold recognition: Alphabets of backbone geometry. Proteins 2003, 51, 504–514. [Google Scholar] [CrossRef]
- Ghouzam, Y.; Postic, G.; Guerin, P.E.; de Brevern, A.G.; Gelly, J.C. ORION: A web server for protein fold recognition and structure prediction using evolutionary hybrid profiles. Sci. Rep. 2016, 6, 28268. [Google Scholar] [CrossRef] [PubMed]
- de Brevern, A.G.; Etchebest, C.; Hazout, S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins 2000, 41, 271–287. [Google Scholar] [CrossRef] [Green Version]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noel, F.; Shinada, N.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.; et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craveur, P.; Narwani, T.J.; Rebehmed, J.; de Brevern, A.G. Investigation of the impact of PTMs on the protein backbone conformation. Amino Acids 2019, 51, 1065–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narwani, T.J.; Craveur, P.; Shinada, N.K.; Floch, A.; Santuz, H.; Vattekatte, A.M.; Srinivasan, N.; Rebehmed, J.; Gelly, J.C.; Etchebest, C.; et al. Discrete analyses of protein dynamics. J. Biomol. Struct. Dyn. 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Goguet, M.; Narwani, T.J.; Petermann, R.; Jallu, V.; de Brevern, A.G. In silico analysis of Glanzmann variants of Calf-1 domain of alphaIIbbeta3 integrin revealed dynamic allosteric effect. Sci. Rep. 2017, 7, 8001. [Google Scholar] [CrossRef] [Green Version]
- Craveur, P.; Gres, A.T.; Kirby, K.A.; Liu, D.; Hammond, J.A.; Deng, Y.; Forli, S.; Goodsell, D.S.; Williamson, J.R.; Sarafianos, S.G.; et al. Novel Intersubunit Interaction Critical for HIV-1 Core Assembly Defines a Potentially Targetable Inhibitor Binding Pocket. mBio 2019, 10, e02858-18. [Google Scholar] [CrossRef] [Green Version]
- Ladislav, M.; Cerny, J.; Krusek, J.; Horak, M.; Balik, A.; Vyklicky, L. The LILI Motif of M3-S2 Linkers Is a Component of the NMDA Receptor Channel Gate. Front. Mol. Neurosci. 2018, 11, 113. [Google Scholar] [CrossRef] [Green Version]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Cracking the folding code. Why do some proteins adopt partially folded conformations, whereas other don’t? FEBS Lett. 2002, 514, 181–183. [Google Scholar] [CrossRef]
- Mitic, N.S.; Malkov, S.N.; Kovacevic, J.J.; Pavlovic-Lazetic, G.M.; Beljanski, M.V. Structural disorder of plasmid-encoded proteins in Bacteria and Archaea. BMC Bioinform. 2018, 19, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toto, A.; Malagrino, F.; Visconti, L.; Troilo, F.; Pagano, L.; Brunori, M.; Jemth, P.; Gianni, S. Templated folding of intrinsically disordered proteins. J. Biol. Chem. 2020, 295, 6586–6593. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Kragelj, J.; Blackledge, M.; Jensen, M.R. Ensemble Calculation for Intrinsically Disordered Proteins Using NMR Parameters. Adv. Exp. Med. Biol. 2015, 870, 123–147. [Google Scholar]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [Green Version]
- Ezerski, J.C.; Zhang, P.; Jennings, N.C.; Waxham, M.N.; Cheung, M.S. Molecular Dynamics Ensemble Refinement of Intrinsically Disordered Peptides According to Deconvoluted Spectra from Circular Dichroism. Biophys. J. 2020, 118, 1665–1678. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.; Chen, J. Targeting Intrinsically Disordered Proteins through Dynamic Interactions. Biomolecules 2020, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, M.F.; Lei, M.; Rader, A.J.; Jacobs, D.J.; Kuhn, L.A. Protein flexibility and dynamics using constraint theory. J. Mol. Graph. Model. 2001, 19, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Carugo, O. Atomic displacement parameters in structural biology. Amino Acids 2018, 50, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder here, there, and everywhere, and nowhere to escape from it. Cell. Mol. Life Sci. 2017, 74, 3065–3067. [Google Scholar] [CrossRef]
- Varadi, M.; Kosol, S.; Lebrun, P.; Valentini, E.; Blackledge, M.; Dunker, A.K.; Felli, I.C.; Forman-Kay, J.D.; Kriwacki, R.W.; Pierattelli, R.; et al. pE-DB: A database of structural ensembles of intrinsically disordered and of unfolded proteins. Nucleic Acids Res. 2014, 42, D326–D335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melarkode Vattekatte, A.; Narwani, T.J.; Floch, A.; Maljkovic, M.; Bisoo, S.; Shinada, N.K.; Kranjc, A.; Gelly, J.C.; Srinivasan, N.; Mitic, N.; et al. A structural entropy index to analyse local conformations in intrinsically disordered proteins. J. Struct. Biol. 2020, 210, 107464. [Google Scholar]
- Melarkode Vattekatte, A.; Narwani, T.J.; Floch, A.; Maljkovic, M.; Bisoo, S.; Shinada, N.K.; Kranjc, A.; Gelly, J.C.; Srinivasan, N.; Mitic, N.; et al. Data set of intrinsically disordered proteins analysed at a local protein conformation level. Data Brief 2020, 29, 105383. [Google Scholar] [CrossRef]
- Mittag, T.; Marsh, J.; Grishaev, A.; Orlicky, S.; Lin, H.; Sicheri, F.; Tyers, M.; Forman-Kay, J.D. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure 2010, 18, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Weeks, S.D.; Baranova, E.V.; Heirbaut, M.; Beelen, S.; Shkumatov, A.V.; Gusev, N.B.; Strelkov, S.V. Molecular structure and dynamics of the dimeric human small heat shock protein HSPB6. J. Struct. Biol. 2014, 185, 342–354. [Google Scholar] [CrossRef]
- Allison, J.R.; Rivers, R.C.; Christodoulou, J.C.; Vendruscolo, M.; Dobson, C.M. A relationship between the transient structure in the monomeric state and the aggregation propensities of alpha-synuclein and beta-synuclein. Biochemistry 2014, 53, 7170–7183. [Google Scholar] [CrossRef]
- Sivakolundu, S.G.; Bashford, D.; Kriwacki, R.W. Disordered p27Kip1 exhibits intrinsic structure resembling the Cdk2/cyclin A-bound conformation. J. Mol. Biol. 2005, 353, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Mertens, H.D.; Piljic, A.; Schultz, C.; Svergun, D.I. Conformational analysis of a genetically encoded FRET biosensor by SAXS. Biophys. J. 2012, 102, 2866–2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacot-Davis, V.R.; Ciomperlik, J.J.; Basta, H.A.; Cornilescu, C.C.; Palmenberg, A.C. Solution structures of Mengovirus Leader protein, its phosphorylated derivatives, and in complex with nuclear transport regulatory protein, RanGTPase. Proc. Natl. Acad. Sci. USA 2014, 111, 15792–15797. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martinez, M.; Crehuet, R. Application of the maximum entropy principle to determine ensembles of intrinsically disordered proteins from residual dipolar couplings. Phys. Chem. Chem. Phys. 2014, 16, 26030–26039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterckx, Y.G.; Volkov, A.N.; Vranken, W.F.; Kragelj, J.; Jensen, M.R.; Buts, L.; Garcia-Pino, A.; Jove, T.; Van Melderen, L.; Blackledge, M.; et al. Small-angle X-ray scattering- and nuclear magnetic resonance-derived conformational ensemble of the highly flexible antitoxin PaaA2. Structure 2014, 22, 854–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Biasio, A.; Ibanez de Opakua, A.; Cordeiro, T.N.; Villate, M.; Merino, N.; Sibille, N.; Lelli, M.; Diercks, T.; Bernado, P.; Blanco, F.J. p15PAF is an intrinsically disordered protein with nonrandom structural preferences at sites of interaction with other proteins. Biophys. J. 2014, 106, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Ozenne, V.; Schneider, R.; Yao, M.; Huang, J.R.; Salmon, L.; Zweckstetter, M.; Jensen, M.R.; Blackledge, M. Mapping the potential energy landscape of intrinsically disordered proteins at amino acid resolution. J. Am. Chem. Soc. 2012, 134, 15138–15148. [Google Scholar] [CrossRef] [Green Version]
- Marsh, J.A.; Forman-Kay, J.D. Structure and disorder in an unfolded state under nondenaturing conditions from ensemble models consistent with a large number of experimental restraints. J. Mol. Biol. 2009, 391, 359–374. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.P.; Agarwal, G.; Mahajan, S.; Gelly, J.-C.; Swapna, L.S.; Offmann, B.; Cadet, F.; Bornot, A.; Tyagi, M.; Valadié, H.; et al. A short survey on Protein Blocks. Biophys. Rev. 2010, 2, 137–145. [Google Scholar] [CrossRef]
- Jallu, V.; Poulain, P.; Fuchs, P.F.; Kaplan, C.; de Brevern, A.G. Modeling and molecular dynamics simulations of the V33 variant of the integrin subunit beta3: Structural comparison with the L33 (HPA-1a) and P33 (HPA-1b) variants. Biochimie 2014, 105, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Barnoud, J.; Santuz, H.; Craveur, P.; Joseph, A.P.; Jallu, V.; de Brevern, A.G.; Poulain, P. PBxplore: A tool to analyze local protein structure and deformability with Protein Blocks. PeerJ 2017, 5, e4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Erdos, G.; Dosztanyi, Z. Analyzing Protein Disorder with IUPred2A. Curr. Protoc. Bioinform. 2020, 70, e99. [Google Scholar] [CrossRef] [Green Version]
- Python Software Foundation. Available online: https://www.python.org/ (accessed on 14 June 2020).
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2017. Available online: https://www.R-project.org/ (accessed on 14 June 2020).
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.7.2.2; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- DeLano, W.L.T. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. Available online: http://www.pymol.org (accessed on 14 June 2020).
- Narwani, T.J.; Craveur, P.; Shinada, N.K.; Santuz, H.; Rebehmed, J.; Etchebest, C.; de Brevern, A.G. Dynamics and deformability of α-, 310- and π-helices. Arch. Biol. Sci. 2018, 70, 21–31. [Google Scholar] [CrossRef]
- Pavlovic-Lazetic, G.M.; Mitic, N.S.; Kovacevic, J.J.; Obradovic, Z.; Malkov, S.N.; Beljanski, M.V. Bioinformatics analysis of disordered proteins in prokaryotes. BMC Bioinform. 2011, 12, 66. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.T.; Mulder, F.A.A. Quality and bias of protein disorder predictors. Sci. Rep. 2019, 9, 5137. [Google Scholar] [CrossRef] [Green Version]
- de Brevern, A.G.; Bornot, A.; Craveur, P.; Etchebest, C.; Gelly, J.C. PredyFlexy: Flexibility and local structure prediction from sequence. Nucleic Acids Res. 2012, 40, W317–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdigao, N.; Rosa, A. Dark Proteome Database: Studies on Dark Proteins. High Throughput 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieutaud, P.; Ferron, F.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. How disordered is my protein and what is its disorder for? A guide through the “dark side” of the protein universe. Intrinsically Disord Proteins 2016, 4, e1259708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Finkelstein, A.V. Life in Phases: Intra- and Inter- Molecular Phase Transitions in Protein Solutions. Biomolecules 2019, 9, 842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. New technologies to analyse protein function: An intrinsic disorder perspective. F1000Res 2020, 9, F1000 Faculty Rev-101. [Google Scholar] [CrossRef] [Green Version]
- Barik, A.; Katuwawala, A.; Hanson, J.; Paliwal, K.; Zhou, Y.; Kurgan, L. DEPICTER: Intrinsic Disorder and Disorder Function Prediction Server. J. Mol. Biol 2020, 432, 3379–3387. [Google Scholar] [CrossRef]
- Blundell, T.L.; Gupta, M.N.; Hasnain, S.E. Intrinsic disorder in proteins: Relevance to protein assemblies, drug design and host-pathogen interactions. Prog. Biophys. Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Nagibina, G.S.; Glukhova, K.A.; Uversky, V.N.; Melnik, T.N.; Melnik, B.S. Intrinsic Disorder-Based Design of Stable Globular Proteins. Biomolecules 2019, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Kim, D.H.; Han, J.J.; Cha, E.J.; Lim, J.E.; Cho, Y.J.; Lee, C.; Han, K.H. Understanding pre-structured motifs (PreSMos) in intrinsically unfolded proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef]
- Kim, D.H.; Han, K.H. PreSMo Target-Binding Signatures in Intrinsically Disordered Proteins. Mol. Cells 2018, 41, 889–899. [Google Scholar]
- Sharma, R.; Sharma, A.; Raicar, G.; Tsunoda, T.; Patil, A. OPAL+: Length-Specific MoRF Prediction in Intrinsically Disordered Protein Sequences. Proteomics 2019, 19, e1800058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Uversky, V.N.; Chen, Z.; Dunker, A.K.; Obradovic, Z. Short Linear Motifs recognized by SH2, SH3 and Ser/Thr Kinase domains are conserved in disordered protein regions. BMC Genom. 2008, 9 (Suppl. 2), S26. [Google Scholar] [CrossRef] [Green Version]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wu, Z.; Oldfield, C.J.; Wang, C.; Kurgan, L. Quality assessment for the putative intrinsic disorder in proteins. Bioinformatics 2019, 35, 1692–1700. [Google Scholar] [CrossRef] [Green Version]
- Katuwawala, A.; Oldfield, C.J.; Kurgan, L. DISOselect: Disorder predictor selection at the protein level. Protein Sci. 2020, 29, 184–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.; Uversky, V.N.; Schnell, S. On the Need to Develop Guidelines for Characterizing and Reporting Intrinsic Disorder in Proteins. Proteomics 2019, 19, e1800415. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Brevern, A.G. Analysis of Protein Disorder Predictions in the Light of a Protein Structural Alphabet. Biomolecules 2020, 10, 1080. https://doi.org/10.3390/biom10071080
de Brevern AG. Analysis of Protein Disorder Predictions in the Light of a Protein Structural Alphabet. Biomolecules. 2020; 10(7):1080. https://doi.org/10.3390/biom10071080
Chicago/Turabian Stylede Brevern, Alexandre G. 2020. "Analysis of Protein Disorder Predictions in the Light of a Protein Structural Alphabet" Biomolecules 10, no. 7: 1080. https://doi.org/10.3390/biom10071080
APA Stylede Brevern, A. G. (2020). Analysis of Protein Disorder Predictions in the Light of a Protein Structural Alphabet. Biomolecules, 10(7), 1080. https://doi.org/10.3390/biom10071080