Protein–Protein Interactions Mediated by Intrinsically Disordered Protein Regions Are Enriched in Missense Mutations

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
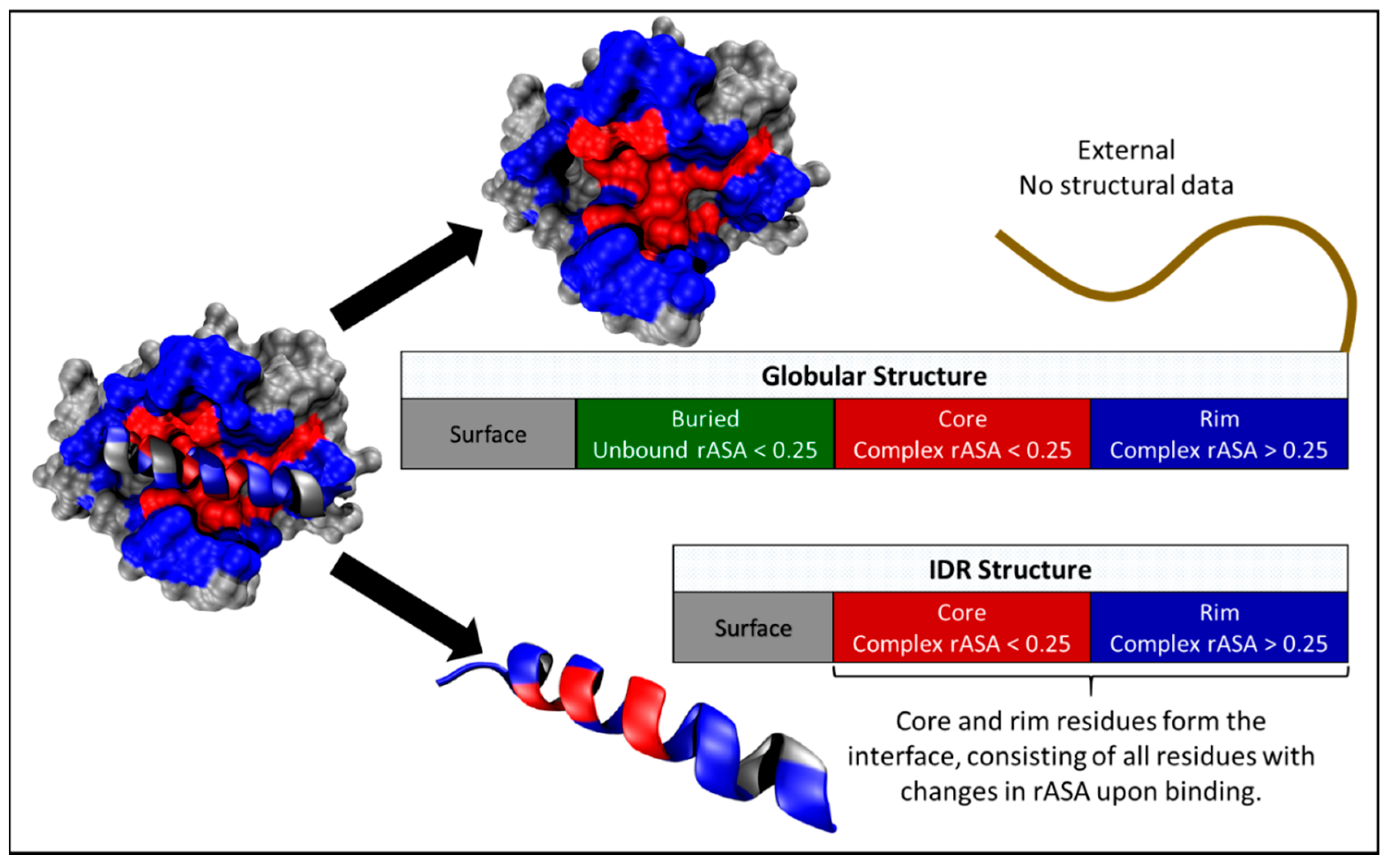
2.1. Structural Data
2.2. Defining Intrinsically Disordered Regions (IDR) Interaction Datasets
2.3. Mapping Mutations to Globular and IDR Interaction Structural Data
2.4. Odds Ratio Calculations
3. Results
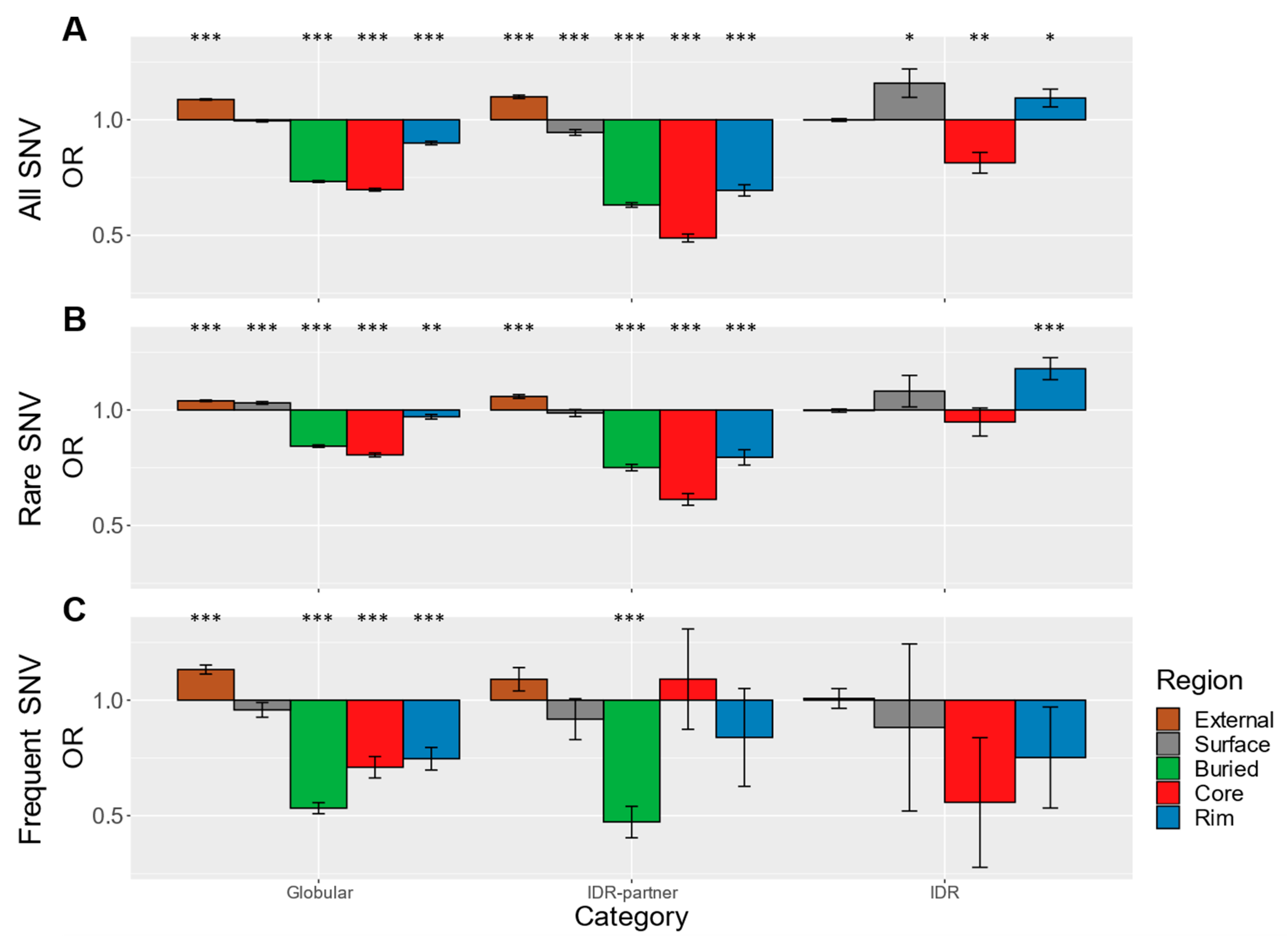
3.1. Disease-Associated Single Nucleotide Variants (SNVs) Are Enriched at IDR Interaction Interfaces
3.2. gnomAD SNVs Are Depleted at IDR Interaction Interfaces
3.3. Robustness of Datasets and Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sahni, N.; Yi, S.; Taipale, M.; Fuxman Bass, J.I.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, A.; Sternberg, M.J.E. The Contribution of Missense Mutations in Core and Rim Residues of Protein-Protein Interfaces to Human Disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savojardo, C.; Babbi, G.; Martelli, P.; Casadio, R. Functional and Structural Features of Disease-Related Protein Variants. Int. J. Mol. Sci. 2019, 20, 1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehr, H.; Jang, S.-H.J.; Duarte, J.M.; Wierling, C.; Lehrach, H.; Lappe, M.; Lange, B.M.H. The structural impact of cancer-associated missense mutations in oncogenes and tumor suppressors. Mol. Cancer 2011, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Moult, J. SNPs, protein structure, and disease. Hum. Mutat. 2001, 17, 263–270. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Razali, R.; Wass, M.N.; Sternberg, M.J.E. Protein-protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum. Mutat. 2012, 33, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Stefl, S.; Nishi, H.; Petukh, M.; Panchenko, A.R.; Alexov, E. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013, 425, 3919–3936. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, P.; Janin, J. Dissecting protein-protein recognition sites. Proteins 2002, 47, 334–343. [Google Scholar] [CrossRef]
- Levy, E.D. A Simple Definition of Structural Regions in Proteins and Its Use in Analyzing Interface Evolution. J. Mol. Biol. 2010, 403, 660–670. [Google Scholar] [CrossRef]
- Wang, X.; Wei, X.; Thijssen, B.; Das, J.; Lipkin, S.M.; Yu, H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat. Biotechnol. 2012, 30, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Tyagi, M.; Teng, S.; Shoemaker, B.A.; Hashimoto, K.; Alexov, E.; Wuchty, S.; Panchenko, A.R. Cancer missense mutations alter binding properties of proteins and their interaction networks. PLoS ONE 2013, 8, e66273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engin, H.B.; Kreisberg, J.F.; Carter, H. Structure-Based Analysis Reveals Cancer Missense Mutations Target Protein Interaction Interfaces. PLoS ONE 2016, 11, e0152929. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.-L.; Li, M.; Goncearenco, A.; Panchenko, A.R. Finding driver mutations in cancer: Elucidating the role of background mutational processes. PLoS Comput. Biol. 2019, 15, e1006981. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sivley, R.M.; Dou, X.; Meiler, J.; Bush, W.S.; Capra, J.A. Comprehensive Analysis of Constraint on the Spatial Distribution of Missense Variants in Human Protein Structures. Am. J. Hum. Genet. 2018, 102, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Saint Pierre, A.; Génin, E. How important are rare variants in common disease? Brief. Funct. Genom. 2014, 13, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-T.; Yeung, C.K.L.; Omland, K.E.; Pang, E.-L.; Hao, Y.; Liao, B.-Y.; Cao, H.-F.; Zhang, B.-W.; Yeh, C.-F.; Hung, C.-M.; et al. Standing genetic variation as the predominant source for adaptation of a songbird. Proc. Natl. Acad. Sci. USA 2019, 116, 2152–2157. [Google Scholar] [CrossRef] [Green Version]
- Key, F.M.; Teixeira, J.C.; de Filippo, C.; André, A.M. Advantageous diversity maintained by balancing selection in humans. Curr. Opin. Genet. Dev. 2014, 29, 45–51. [Google Scholar] [CrossRef]
- Yi, X.; Liang, Y.; Huerta-Sanchez, E.; Jin, X.; Cuo, Z.X.P.; Pool, J.E.; Xu, X.; Jiang, H.; Vinckenbosch, N.; Korneliussen, T.S.; et al. Sequencing of 50 Human Exomes Reveals Adaptation to High Altitude. Science 2010, 329, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Kryukov, G.V.; Pennacchio, L.A.; Sunyaev, S.R. Most rare missense alleles are deleterious in humans: Implications for complex disease and association studies. Am. J. Hum. Genet. 2007, 80, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marouli, E.; Graff, M.; Medina-Gomez, C.; Lo, K.S.; Wood, A.R.; Kjaer, T.R.; Fine, R.S.; Lu, Y.; Schurmann, C.; Highland, H.M.; et al. Rare and low-frequency coding variants alter human adult height. Nature 2017, 542, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A Million peptide motifs for the molecular biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.-H.; Kim, P.M. The present and the future of motif-mediated protein-protein interactions. Curr. Opin. Struct. Biol. 2018, 50, 162–170. [Google Scholar] [CrossRef]
- Le Gall, T.; Romero, P.R.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in the Protein Data Bank. J. Biomol. Struct. Dyn. 2007, 24, 325–342. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.L.; Uversky, V.N. Intrinsic disorder and posttranslational modifications: The darker side of the biological dark matter. Front. Genet. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pentony, M.M.; Jones, D.T. Modularity of intrinsic disorder in the human proteome. Proteins Struct. Funct. Bioinf. 2010, 78, 212–221. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta Rev. Cancer 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [Green Version]
- Mosca, R.; Pache, R.A.; Aloy, P. The role of structural disorder in the rewiring of protein interactions through evolution. Mol. Cell. Proteom. 2012, 11, M111.014969. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006, 2, e100. [Google Scholar] [CrossRef]
- Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Functional analysis of human hub proteins and their interactors involved in the intrinsic disorder-enriched interactions. Int. J. Mol. Sci. 2017, 18, 2761. [Google Scholar] [CrossRef] [Green Version]
- Pajkos, M.; Mészáros, B.; Simon, I.; Dosztányi, Z. Is there a biological cost of protein disorder? Analysis of cancer-associated mutations. Mol. Biosyst. 2012, 8, 296–307. [Google Scholar] [CrossRef]
- Brown, C.J.; Johnson, A.K.; Dunker, A.K.; Daughdrill, G.W. Evolution and disorder. Curr. Opin. Struct. Biol. 2011, 21, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Markwick, P.R.L.; Oldfield, C.J.; Zhao, X.; Haynes, C.; Uversky, V.N.; Iakoucheva, L.M. Disease-Associated Mutations Disrupt Functionally Important Regions of Intrinsic Protein Disorder. PLoS Comput. Biol. 2012, 8, e1002709. [Google Scholar] [CrossRef]
- Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Davey, N.E.; Gibson, T.J. Proteome-wide analysis of human disease mutations in short linear motifs: Neglected players in cancer? Mol. BioSyst. 2014, 10, 2626–2642. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, B.; Tompa, P.; Simon, I.; Dosztányi, Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 2007, 372, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.C.; Na, D.; Gsponer, J. On the importance of polar interactions for complexes containing intrinsically disordered proteins. PLoS Comput. Biol. 2013, 9, e1003192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The Structural Basis of Peptide-Protein Binding Strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.T.C.; Gsponer, J. Predicting Protein–Protein Interfaces that Bind Intrinsically Disordered Protein Regions. J. Mol. Biol. 2019, 431. [Google Scholar] [CrossRef]
- Mottaz, A.; David, F.P.A.; Veuthey, A.-L.; Yip, Y.L. Easy retrieval of single amino-acid polymorphisms and phenotype information using SwissVar. Bioinformatics 2010, 26, 851–852. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Fuxreiter, M. The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 2016, 165, 1055–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deiana, A.; Forcelloni, S.; Porrello, A.; Giansanti, A. Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS ONE 2019, 14, e0217889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anbo, H.; Sato, M.; Okoshi, A.; Fukuchi, S. Functional Segments on Intrinsically Disordered Regions in Disease-Related Proteins. Biomolecules 2019, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef]
- Mitternacht, S. FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Research 2016, 5, 189. [Google Scholar] [CrossRef]
- Magrane, M.; Consortium, U. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, D.; Tabaro, F.; Paladin, L.; Necci, M.; Mičetić, I.; Camilloni, C.; Davey, N.; Dosztányi, Z.; Mészáros, B.; Monzon, A.M.; et al. MobiDB 3.0: More annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018, 46, D471–D476. [Google Scholar] [CrossRef]
- Gouw, M.; Michael, S.; Sámano-Sánchez, H.; Kumar, M.; Zeke, A.; Lang, B.; Bely, B.; Chemes, L.B.; Davey, N.E.; Deng, Z.; et al. The eukaryotic linear motif resource—2018 update. Nucleic Acids Res. 2018, 46, D428–D434. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Vitkup, D.; Sander, C.; Church, G.M. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003, 4, R72. [Google Scholar] [CrossRef] [Green Version]
- McFarland, C.D.; Korolev, K.S.; Kryukov, G.V.; Sunyaev, S.R.; Mirny, L.A. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. USA 2013, 110, 2910–2915. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.-C.; Chung, S.S.; Fornili, A.; Fraternali, F. Anatomy of protein disorder, flexibility and disease-related mutations. Front. Mol. Biosci. 2015, 2, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, H.; Nakata, J.; Kinoshita, K. Distribution of single-nucleotide variants on protein-protein interaction sites and its relationship with minor allele frequency. Protein Sci. 2016, 25, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Forcelloni, S.; Giansanti, A. Evolutionary Forces and Codon Bias in Different Flavors of Intrinsic Disorder in the Human Proteome. J. Mol. Evol. 2020, 88, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Douglas, G.M.; Patel, P.; Nguyen Ba, A.N.; Moses, A.M. Polymorphism Analysis Reveals Reduced Negative Selection and Elevated Rate of Insertions and Deletions in Intrinsically Disordered Protein Regions. Genome Biol. Evol. 2015, 7, 1815–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fold or not to fold upon binding—Does it really matter? Curr. Opin. Struct. Biol. 2018, 54, 19–25. [Google Scholar] [CrossRef]
- Miskei, M.; Antal, C.; Fuxreiter, M. FuzDB: Database of fuzzy complexes, a tool to develop stochastic structure-function relationships for protein complexes and higher-order assemblies. Nucleic Acids Res. 2017, 45, D228–D235. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fuzziness in Protein Interactions-A Historical Perspective. J. Mol. Biol. 2018, 430, 2278–2287. [Google Scholar] [CrossRef]
- Malhis, N.; Wong, E.T.C.; Nassar, R.; Gsponer, J. Computational identification of MoRFs in protein sequences using Hierarchical application of bayes rule. PLoS ONE 2015, 10, e0141603. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Fornili, A.; Pandini, A.; Lu, H.-C.; Fraternali, F. Specialized Dynamical Properties of Promiscuous Residues Revealed by Simulated Conformational Ensembles. J. Chem. Theory Comput. 2013, 9, 5127–5147. [Google Scholar] [CrossRef]
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein-protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Yadahalli, S.; Li, J.; Lane, D.P.; Gosavi, S.; Verma, C.S. Characterizing the conformational landscape of MDM2-binding p53 peptides using Molecular Dynamics simulations. Sci. Rep. 2017, 7, 15600. [Google Scholar] [CrossRef] [Green Version]
- Dincer, C.; Kaya, T.; Keskin, O.; Gursoy, A.; Tuncbag, N. 3D spatial organization and network-guided comparison of mutation profiles in Glioblastoma reveals similarities across patients. PLoS Comput. Biol. 2019, 15, e1006789. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Kirchner, M.; Uyar, B.; Cheng, J.-Y.; Russo, G.; Hernandez-Miranda, L.R.; Szymborska, A.; Zauber, H.; Rudolph, I.-M.; Willnow, T.E.; et al. Mutations in Disordered Regions Can Cause Disease by Creating Dileucine Motifs. Cell 2018, 175, 239–253.e17. [Google Scholar] [CrossRef] [Green Version]
- Reimand, J.; Bader, G.D. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 2013, 9, 637. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Lawrence, M.S.; Polak, P.; Leshchiner, I.; Lage, K.; Golub, T.R.; Lander, E.S.; Getz, G. Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc. Natl. Acad. Sci. USA 2015, 112, E5486–E5495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhuzimi, E.; Leal, L.G.; Sternberg, M.J.E.; David, A. Properties of human genes guided by their enrichment in rare and common variants. Hum. Mutat. 2018, 39, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Tokuriki, N.; Tawfik, D.S. Protein dynamism and evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlich, Y.; Reeb, J.; Hecht, M.; Schelling, M.; De Beer, T.A.P.; Bromberg, Y.; Rost, B. Common sequence variants affect molecular function more than rare variants? Sci. Rep. 2017, 7, 1608. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.M.; Sboner, A.; Xia, Y.; Gerstein, M. The role of disorder in interaction networks: A structural analysis. Mol. Syst. Biol. 2008, 4, 179. [Google Scholar] [CrossRef]
- London, N.; Raveh, B.; Schueler-Furman, O. Peptide docking and structure-based characterization of peptide binding: From knowledge to know-how. Curr. Opin. Struct. Biol. 2013, 23, 894–902. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, E.T.C.; So, V.; Guron, M.; Kuechler, E.R.; Malhis, N.; Bui, J.M.; Gsponer, J. Protein–Protein Interactions Mediated by Intrinsically Disordered Protein Regions Are Enriched in Missense Mutations. Biomolecules 2020, 10, 1097. https://doi.org/10.3390/biom10081097
Wong ETC, So V, Guron M, Kuechler ER, Malhis N, Bui JM, Gsponer J. Protein–Protein Interactions Mediated by Intrinsically Disordered Protein Regions Are Enriched in Missense Mutations. Biomolecules. 2020; 10(8):1097. https://doi.org/10.3390/biom10081097
Chicago/Turabian StyleWong, Eric T. C., Victor So, Mike Guron, Erich R. Kuechler, Nawar Malhis, Jennifer M. Bui, and Jörg Gsponer. 2020. "Protein–Protein Interactions Mediated by Intrinsically Disordered Protein Regions Are Enriched in Missense Mutations" Biomolecules 10, no. 8: 1097. https://doi.org/10.3390/biom10081097
APA StyleWong, E. T. C., So, V., Guron, M., Kuechler, E. R., Malhis, N., Bui, J. M., & Gsponer, J. (2020). Protein–Protein Interactions Mediated by Intrinsically Disordered Protein Regions Are Enriched in Missense Mutations. Biomolecules, 10(8), 1097. https://doi.org/10.3390/biom10081097