Predicted Hotspot Residues Involved in Allosteric Signal Transmission in Pro-Apoptotic Peptide—Mcl1 Complexes





Abstract
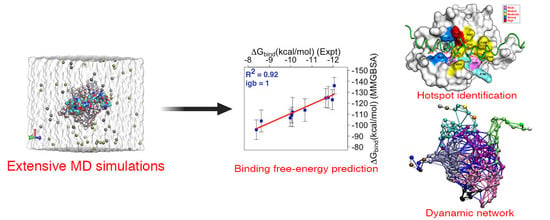
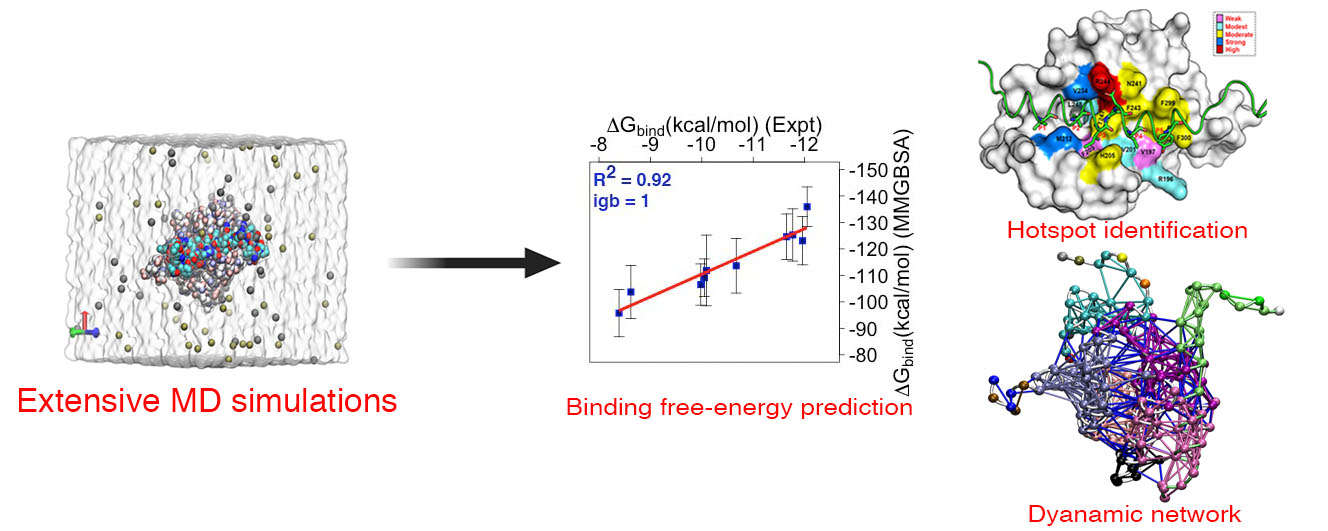
:
1. Introduction
2. Materials and Methods
2.1. Starting Structures Preparation
2.2. System Setup and MD Simulations
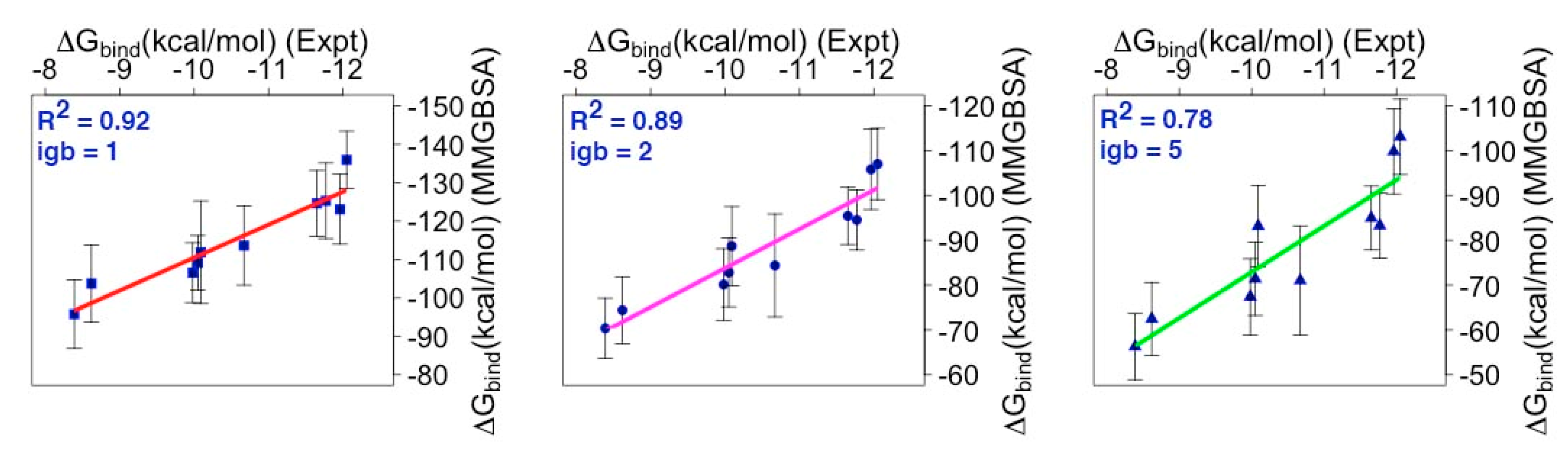
2.3. Binding Free Energy (BFE) Estimation for mMcl1—BH3 Peptide Complexes
2.4. Per Residue Decomposition (PRD) Analysis for mMcl-1—PAP Complexes
2.5. Trajectory Analysis
2.6. Dynamic Network Analysis
3. Results and Discussion
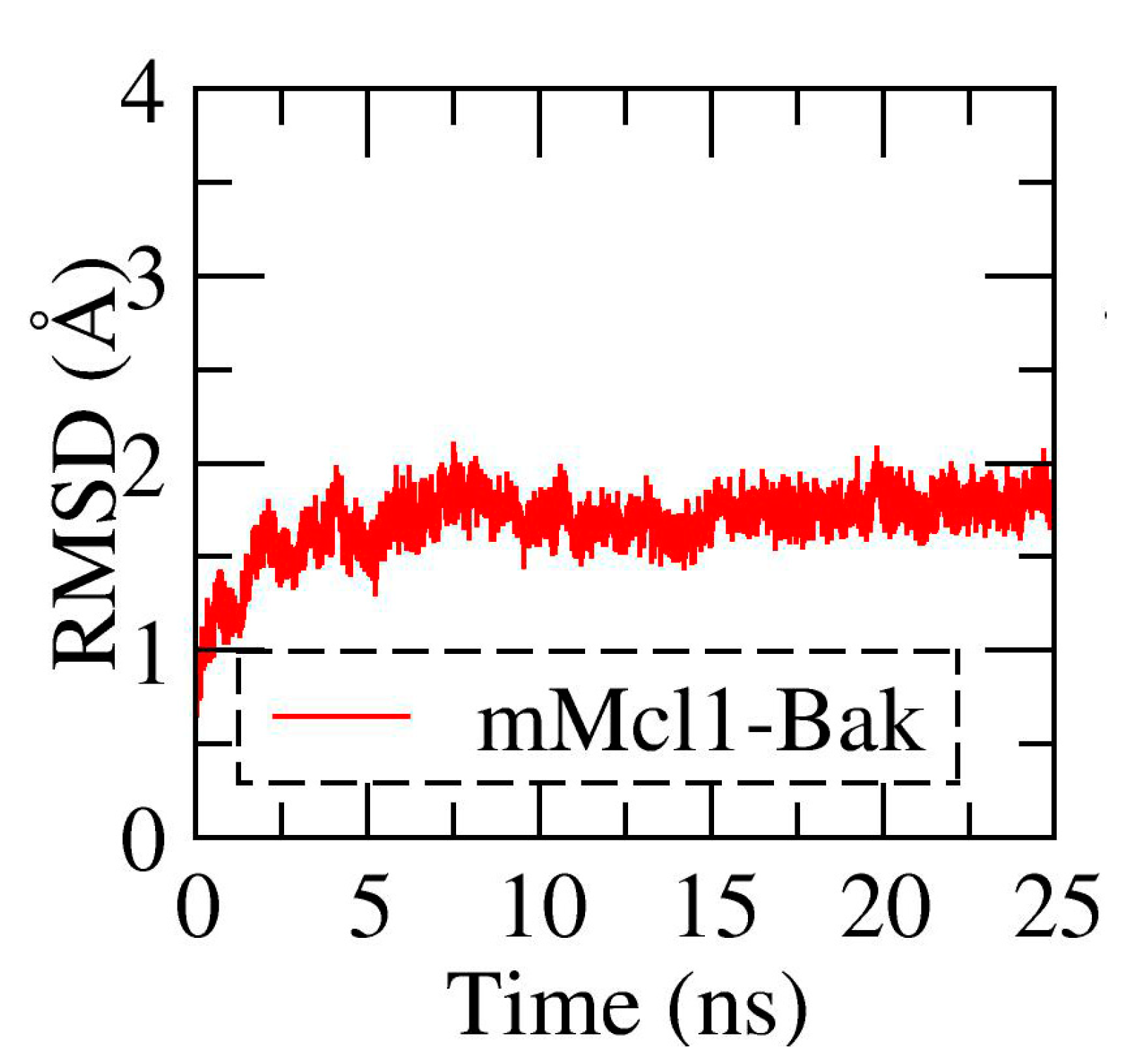
3.1. Conformational Stability Investigation of mMcl1—PAP Complexes during MD Simulations
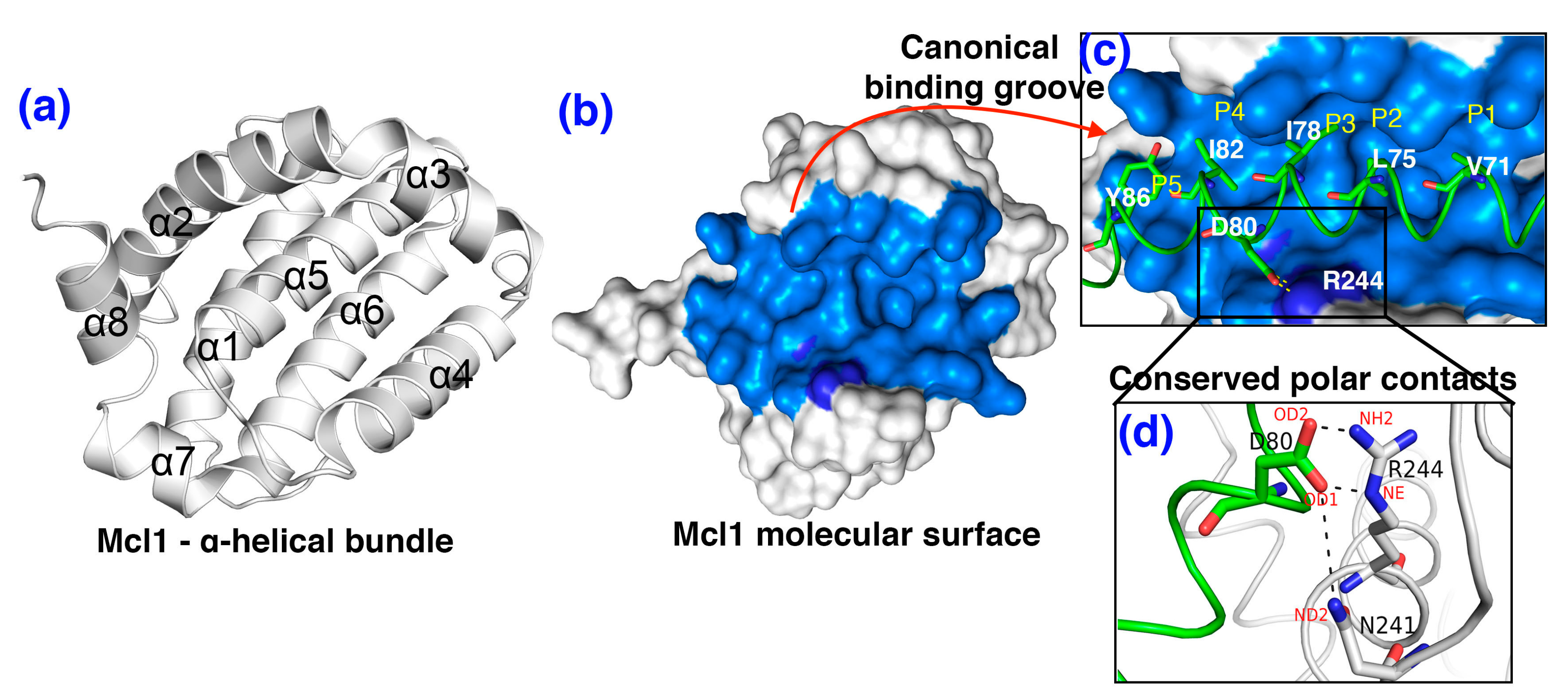
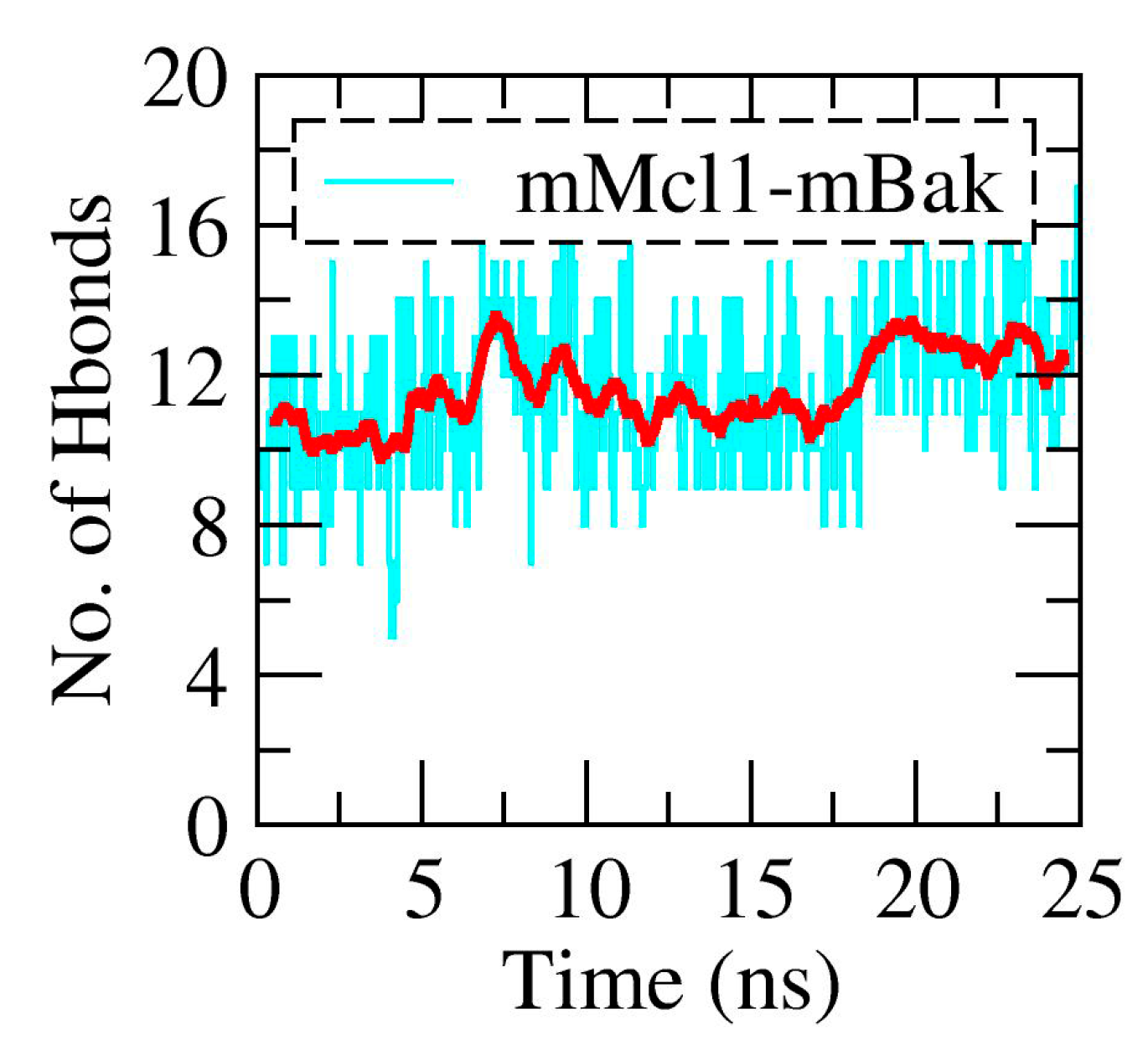
3.2. The Polar Contacts Estimation at the mMcl1—PAP Interfaces During MD Simulations
3.3. Energy Contributions Responsible for mMcl1—PAPs Heterodimerization
3.4. Energy Contributions from the Individual Components Responsible for mMcl1—PAPs Hetero-Dimerization
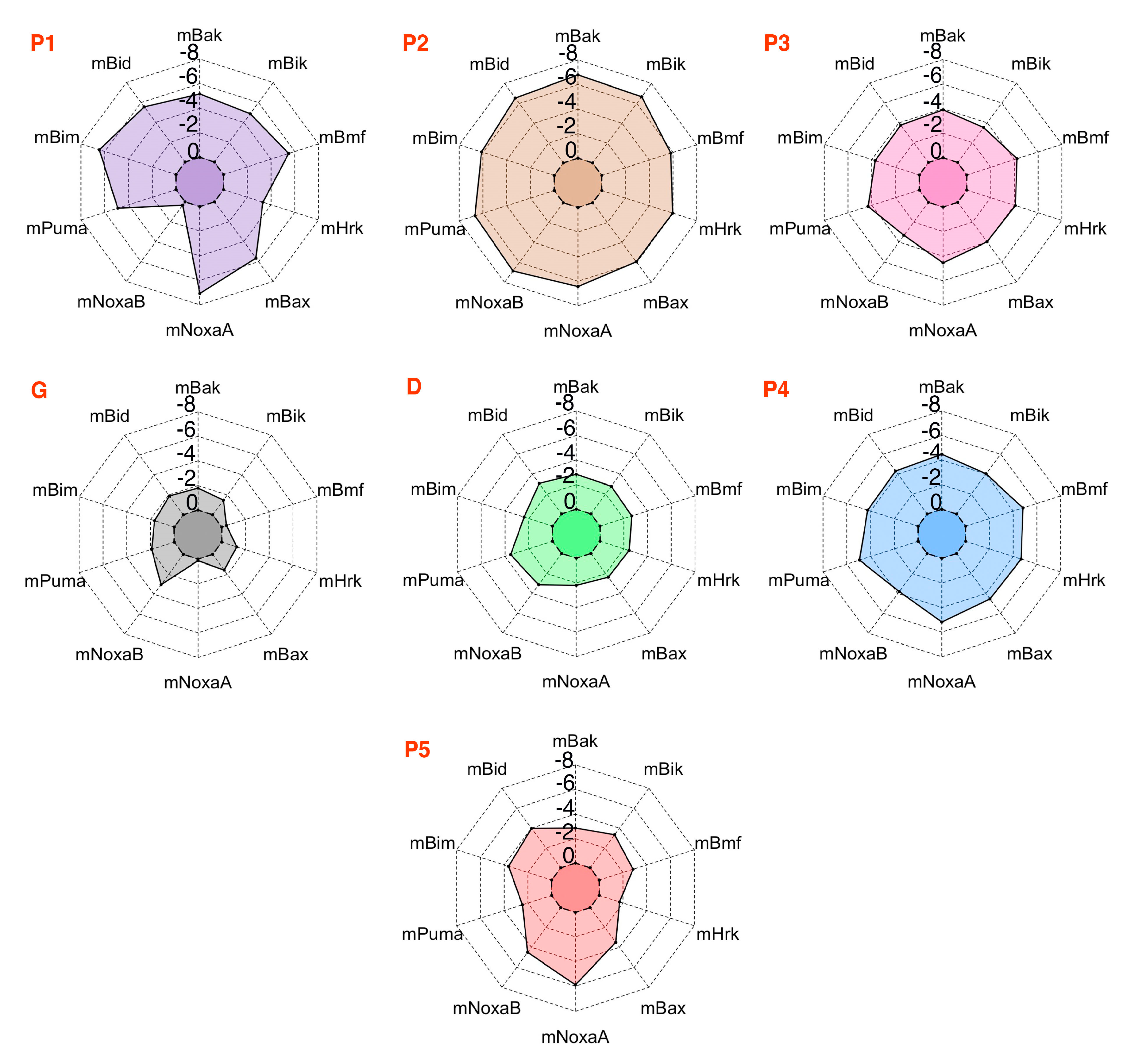
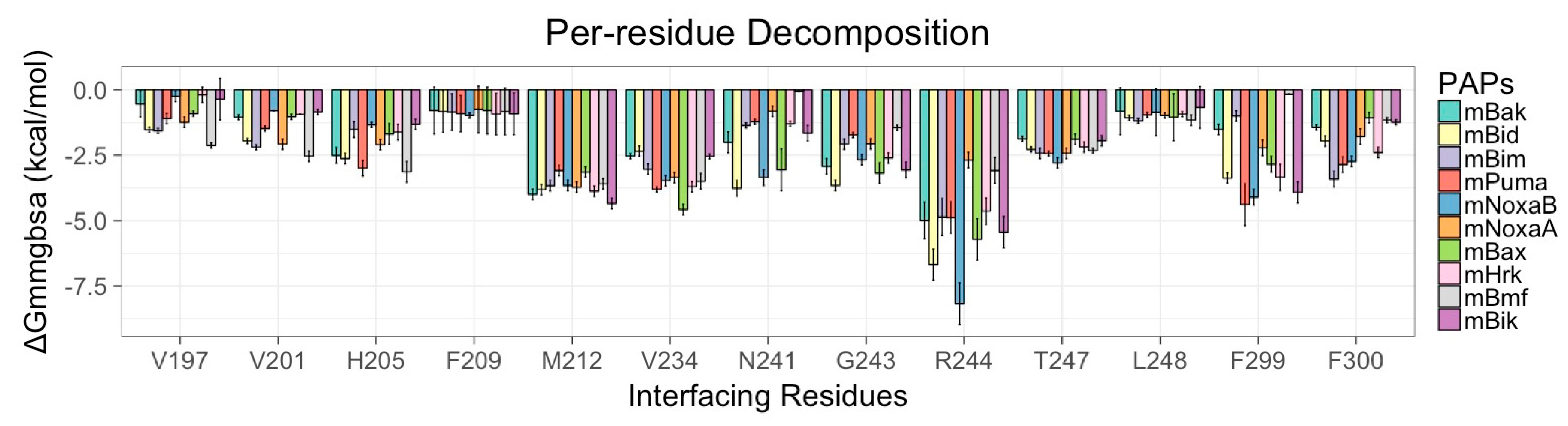
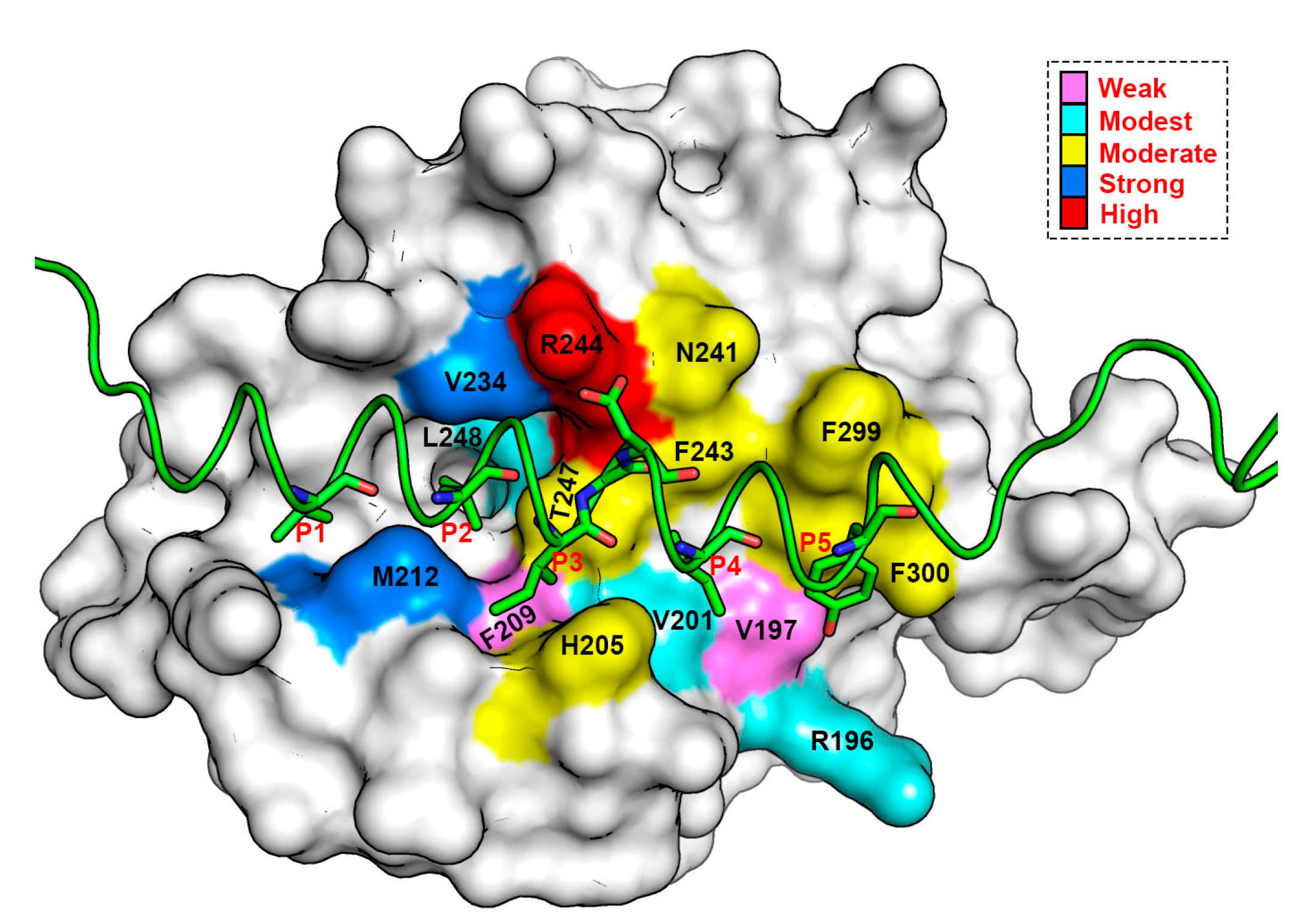
3.5. Energy Contributions from PAPs Key Residues Responsible for Heterodimerization
3.6. Energy Contributions from mMcl1 Key Residues Responsible for Heterodimerization
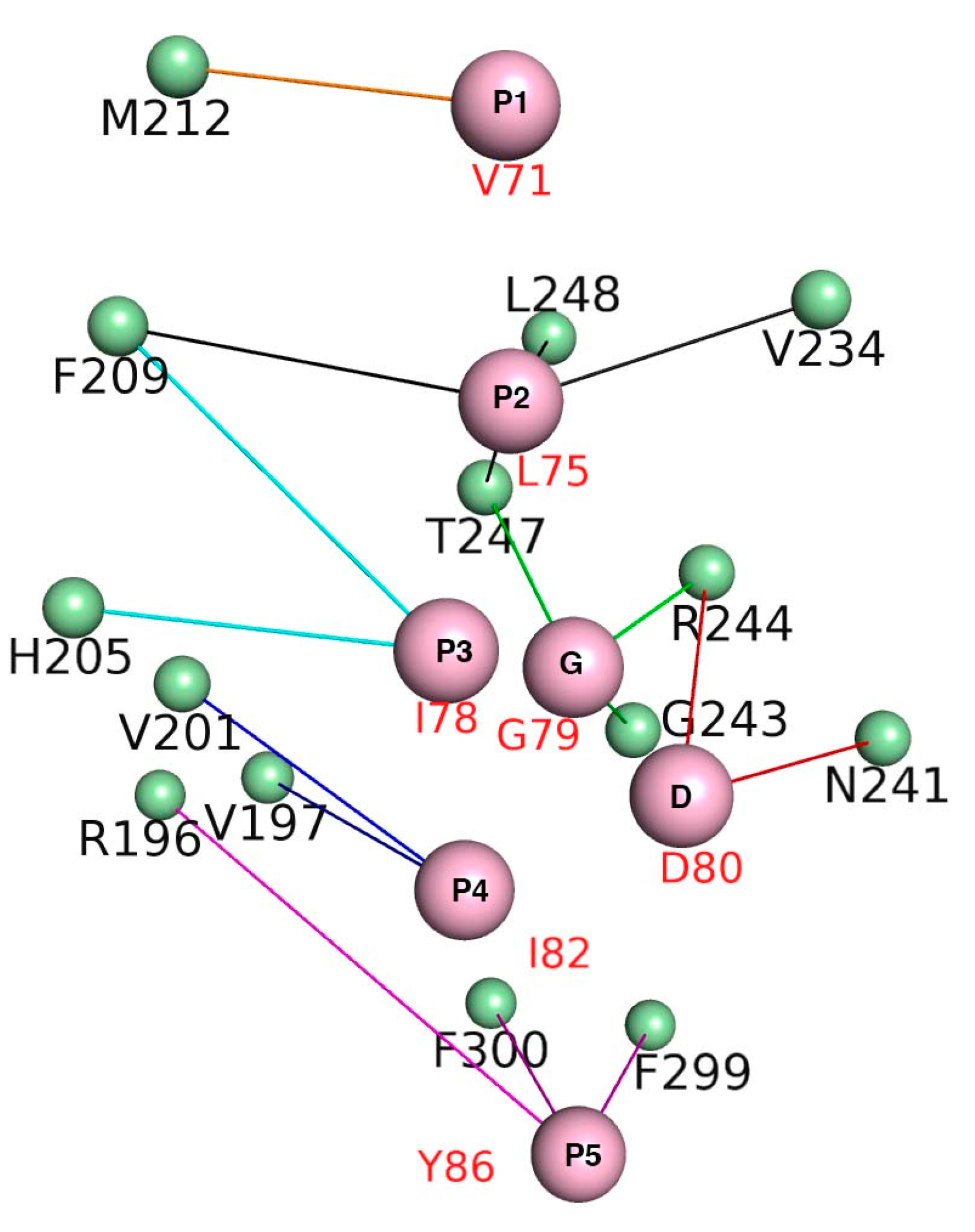
3.7. Crucial Residues Involved in Allosteric Signal Transmission (AST) in mMcl1—PAPs Complexes
3.8. Cross-Correlation Analysis to Investigate Collective Internal Dynamics of mMcl1–Bak Complex
3.9. Community Network Analysis to Investigate Collective Internal Dynamics of mMcl1—Bak Complex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Bcl2 | B-cell lymphoma 2 |
| Mcl1 | Myeloid cell leukemia |
| BH3 | Bcl-2 homology |
| AAP | Anti-apoptotic proteins |
| PAP | Pro-apoptotic proteins |
| BFE | Binding free energy |
| PRD | Per–residue decomposition |
References
- Marsden, V.S.; Strasser, A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 2003, 21, 71–105. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Petros, A.M.; Olejniczak, E.T.; Fesik, S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 2004, 1644, 83–94. [Google Scholar] [CrossRef]
- Lee, J.U.; Hosotani, R.; Wada, M.; Doi, R.; Kosiba, T.; Fujimoto, K.; Miyamoto, Y.; Tsuji, S.; Nakajima, S.; Nishimura, Y.; et al. Role of Bcl-2 family proteins (Bax, Bcl-2 and Bcl-X) on cellular susceptibility to radiation in pancreatic cancer cells. Eur. J. Cancer 1999, 35, 1374–1380. [Google Scholar] [CrossRef]
- Rinkenberger, J.L.; Horning, S.; Klocke, B.; Roth, K.; Korsmeyer, S.J. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000, 14, 23–27. [Google Scholar]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef]
- Opferman, J.T. Unraveling MCL-1 degradation. Cell Death Differ. 2006, 13, 1260–1262. [Google Scholar] [CrossRef]
- Quinn, B.A.; Dash, R.; Azab, B.; Sarkar, S.; Das, S.K.; Kumar, S.; Oyesanya, R.A.; Dasgupta, S.; Dent, P.; Grant, S.; et al. Targeting Mcl-1 for the therapy of cancer. Expert Opin. Investig. Drugs 2011, 20, 1397–1411. [Google Scholar] [CrossRef] [Green Version]
- Michels, J.; Johnson, P.W.; Packham, G. Mcl-1. Int. J. Biochem. Cell Biol. 2005, 37, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.L.; Fire, E.; Keating, A.E.; Walensky, L.D. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol. 2010, 6, 595–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Yap, J.L.; Newell-Rogers, M.K.; Peddaboina, C.; Jiang, W.; Papaconstantinou, H.T.; Jupitor, D.; Rai, A.; Jung, K.Y.; Tubin, R.P.; et al. The novel BH3 alpha-helix mimetic JY-1-106 induces apoptosis in a subset of cancer cells (lung cancer, colon cancer and mesothelioma) by disrupting Bcl-xL and Mcl-1 protein-protein interactions with Bak. Mol. Cancer 2013, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta 2011, 1813, 508–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araujo, D.A.; Lim, J.; Wu, K.C.; Xiang, Y.; Good, A.C.; Skerlj, R.; Fairlie, D.P. Bicyclic Helical Peptides as Dual Inhibitors Selective for Bcl2A1 and Mcl-1 Proteins. J. Med. Chem. 2018, 61, 2962–2972. [Google Scholar] [CrossRef] [Green Version]
- Foight, G.W.; Ryan, J.A.; Gulla, S.V.; Letai, A.; Keating, A.E. Designed BH3 Peptides with High Affinity and Specificity for Targeting Mcl-1 in Cells. ACS Chem. Biol. 2014, 9, 1962–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placzek, W.J.; Sturlese, M.; Wu, B.; Cellitti, J.F.; Wei, J.; Pellecchia, M. Identification of a novel Mcl-1 protein binding motif. J. Biol. Chem. 2011, 286, 39829–39835. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, C.; Li, X.; Song, T.; Wu, Z.; Liang, X.; Zhao, Y.; Shen, X.; Chen, H. Fragment-based design, synthesis, and biological evaluation of N-substituted-5-(4-isopropylthiophenol)-2-hydroxynicotinamide derivatives as novel Mcl-1 inhibitors. Eur. J. Med. Chem. 2013, 60, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, G.; Xie, F.; Song, T.; Chang, X. 3-Thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (S1) based molecules as potent, dual inhibitors of B-cell lymphoma 2 (Bcl-2) and myeloid cell leukemia sequence 1 (Mcl-1): Structure-based design and structure-activity relationship studies. J. Med. Chem. 2011, 54, 1101–1105. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, H.; Wu, G.; Li, Z.; Song, T.; Li, X.Q. Probing the difference between BH3 groove of Mcl-1 and Bcl-2 protein: Implications for dual inhibitors design. Eur. J. Med. Chem. 2011, 46, 3909–3916. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lee, E.F.; van Delft, M.F.; Day, C.L.; Smith, B.J.; Huang, D.C.; Fairlie, W.D.; Hinds, M.G.; Colman, P.M. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6217–6222. [Google Scholar] [CrossRef] [Green Version]
- Ku, B.; Liang, C.Y.; Jung, J.U.; Oh, B.H. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011, 21, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Day, C.L.; Smits, C.; Fan, F.C.; Lee, E.F.; Fairlie, W.D.; Hinds, M.G. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J. Mol. Biol. 2008, 380, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, C.; Czabotar, P.E.; Hinds, M.G.; Day, C.L. Structural plasticity underpins promiscuous binding of the prosurvival protein A1. Structure 2008, 16, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Dai, S.; Zhu, Y.; Marrack, P.; Kappler, J.W. The structure of a Bcl-xL/Bim fragment complex: Implications for Bim function. Immunity 2003, 19, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Muppidi, A.; Doi, K.; Edwardraja, S.; Drake, E.J.; Gulick, A.M.; Wang, H.G.; Lin, Q. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J. Am. Chem. Soc. 2012, 134, 14734–14737. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Botello-Smith, W.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 16; University of California: San Francisco, CA, USA, 15 April 2016. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaer, J.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Chem. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, D.S.; Duke, R.E.; Darden, T.A.; Lybrand, T.P. Staggered Mesh Ewald: An extension of the Smooth Particle-Mesh Ewald method adding great versatility. J. Chem. Theory Comput. 2009, 5, 2322–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Tsui, V.; Case, D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Hamed, M.Y.; Arya, G. Zinc Finger Protein Binding to DNA: An Energy Perspective Using Molecular Dynamics Simulation and Free Energy Calculations on Mutants of both Zinc Finger Domains and their Specific DNA bases. J. Biomol. Struct. Dyn. 2015, 34, 1–41. [Google Scholar] [CrossRef]
- Niinivehmas, S.P.; Virtanen, S.I.; Lehtonen, J.V.; Postila, P.A.; Pentikainen, O.T. Comparison of virtual high-throughput screening methods for the identification of phosphodiesterase-5 inhibitors. J. Chem. Inf. Model. 2011, 51, 1353–1363. [Google Scholar] [CrossRef]
- Wan, H.; Chang, S.; Hu, J.P.; Tian, Y.X.; Tian, X.H. Molecular Dynamics Simulations of Ternary Complexes: Comparisons of LEAFY Protein Binding to Different DNA Motifs. J. Chem. Inf. Model. 2015, 55, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Ylilauri, M.; Pentikainen, O.T. MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Janežič, D.; Karplus, M. Harmonic analysis of large systems. I. Methodology. J. Comput. Chem. 1995, 16, 1522–1542. [Google Scholar] [CrossRef]
- Nogueira, J.J.; González, L. Molecular Dynamics Simulations of Binding Modes between Methylene Blue and DNA with Alternating GC and AT Sequences. Biochemistry 2014, 53, 2391–2412. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hou, T. Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J. Chem. Inf. Model. 2012, 52, 1199–1212. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Senthilkumar, R.; Marimuthu, P.; Paul, P.; Manojkumar, Y.; Arunachalam, S.; Eriksson, J.E.; Johnson, M.S. Plasma Protein Binding of Anisomelic Acid: Spectroscopy and Molecular Dynamic Simulations. J. Chem. Inf. Model. 2016, 56, 2401–2412. [Google Scholar] [CrossRef]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef]
- Hou, T.; Chen, K.; McLaughlin, W.A.; Lu, B.; Wang, W. Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain. PLoS Comput. Biol. 2006, 2, 46–55. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Zhu, T.; Zhang, Q.; Zhang, J.Z. A Comparative Insight into Amprenavir Resistance of Mutations V32I, G48V, I50V, I54V, and I84V in HIV-1 Protease Based on Thermodynamic Integration and MM-PBSA Methods. J. Chem. Inf. Model. 2015, 55, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, D.; Liu, M.; Knabe, W.E.; Yuan, C.; Zhou, D.; Huang, M.; Meroueh, S.O. Small Molecules Engage Hot Spots through Cooperative Binding To Inhibit a Tight Protein-Protein Interaction. Biochemistry 2017, 56, 1768–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Eargle, J.; Luthey-Schulten, Z. NetworkView: 3D display and analysis of protein.RNA interaction networks. Bioinformatics 2012, 28, 3000–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [Green Version]
- Koshy, C.; Parthiban, M.; Sowdhamini, R. 100 ns molecular dynamics simulations to study intramolecular conformational changes in Bax. J. Biomol. Struct. Dyn. 2010, 28, 71–83. [Google Scholar] [CrossRef]
- Lama, D.; Sankararamakrishnan, R. Molecular dynamics simulations of pro-apoptotic BH3 peptide helices in aqueous medium: Relationship between helix stability and their binding affinities to the anti-apoptotic protein Bcl-X-L. J. Comput. Aided Mol. Des. 2011, 25, 413–426. [Google Scholar] [CrossRef]
- Modi, V.; Lama, D.; Sankararamakrishnan, R. Relationship between helix stability and binding affinities: Molecular dynamics simulations of Bfl-1/A1-binding pro-apoptotic BH3 peptide helices in explicit solvent. J. Biomol. Struct. Dyn. 2013, 31, 65–77. [Google Scholar] [CrossRef]
- Marimuthu, P.; Singaravelu, K. Deciphering the crucial residues involved in heterodimerization of Bak peptide and anti-apoptotic proteins for apoptosis. J. Biomol. Struct. Dyn. 2018, 36, 1637–1648. [Google Scholar] [CrossRef]
- Marimuthu, P.; Balasubramanian, P.K.; Singaravelu, K. Deciphering the crucial molecular properties of a series of Benzothiazole Hydrazone inhibitors that targets anti-apoptotic Bcl-xL protein. J. Biomol. Struct. Dyn. 2018, 36, 2654–2667. [Google Scholar] [CrossRef]
- Marimuthu, P.; Singaravelu, K. Unraveling the Molecular Mechanism of Benzothiophene and Benzofuran scaffold merged compounds binding to anti-apoptotic Myeloid cell leukemia 1. J. Biomol. Struct. Dyn. 2019, 37, 1992–2003. [Google Scholar] [CrossRef]
- Singaravelu, K.; Balasubramanian, P.K.; Marimuthu, P. Investigating the Molecular Basis of N-Substituted 1-Hydroxy-4-Sulfamoyl-2-Naphthoate Compounds Binding to Mcl1. Processes 2019, 7, 224. [Google Scholar] [CrossRef] [Green Version]
- Marimuthu, P.; Singaravelu, K. Prediction of Hot Spots at Myeloid Cell Leukemia-1-Inhibitors Interface using Energy Estimation and Alanine Scanning Mutagenesis. Biochemistry 2018, 57, 1249–1261. [Google Scholar] [CrossRef] [Green Version]
- Marimuthu, P.; Razzokov, J.; Eshonqulov, G. Disruption of conserved polar interactions causes a sequential release of Bim mutants from the canonical binding groove of Mcl1. Int. J. Biol. Macromol. 2020, 158, 364–374. [Google Scholar] [CrossRef]
- Lama, D.; Sankararamakrishnan, R. Anti-apoptotic Bcl-XL protein in complex with BH3 peptides of pro-apoptotic Bak, Bad, and Bim proteins: Comparative molecular dynamics simulations. Proteins 2008, 73, 492–514. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Thomas, L.L. Perspective on Free-Energy Perturbation Calculations for Chemical Equilibria. J. Chem. Theory Comput. 2008, 4, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Fratev, F.; Sirimulla, S. An Improved Free Energy Perturbation FEP+ Sampling Protocol for Flexible Ligand-Binding Domains. Sci. Rep. 2019, 9, 16829. [Google Scholar] [CrossRef] [Green Version]
- Shyu, C.; Ytreberg, F.M. Reducing the bias and uncertainty of free energy estimates by using regression to fit thermodynamic integration data. J. Comput. Chem. 2009, 30, 2297–2304. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Virtanen, S.I.; Niinivehmas, S.P.; Pentikainen, O.T. Case-specific performance of MM-PBSA, MM-GBSA, and SIE in virtual screening. J. Mol. Graph. Model. 2015, 62, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boersma, M.D.; Sadowsky, J.D.; Tomita, Y.A.; Gellman, S.H. Hydrophile scanning as a complement to alanine scanning for exploring and manipulating protein-protein recognition: Application to the Bim BH3 domain. Protein Sci. 2008, 17, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Zhou, H.; Dong, Z.; Tao, P. Recognition of protein allosteric states and residues: Machine learning approaches. J. Comput. Chem. 2018, 39, 1481–1490. [Google Scholar] [CrossRef]
- Yoon, H.J.; Lee, S.; Park, S.J.; Wu, S. Network approach of the conformational change of c-Src, a tyrosine kinase, by molecular dynamics simulation. Sci. Rep. 2018, 8, 5673. [Google Scholar] [CrossRef]
- Day, C.L.; Chen, L.; Richardson, S.J.; Harrison, P.J.; Huang, D.C.; Hinds, M.G. Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J. Biol. Chem. 2005, 280, 4738–4744. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | KD (nM) | ΔGbind | igb = 1 | igb = 2 | igb = 5 |
|---|---|---|---|---|---|
| mBak | 1.33 | −12.05 | −135.91 ± 0.39 | −107.01 ± 0.35 | −103.07 ± 0.37 |
| mBid | 1.55 | −11.96 | −132.66 ± 0.45 | −105.79 ± 0.40 | −99.81 ± 0.42 |
| mBim | 2.16 | −11.77 | −125.26 ± 0.34 | −94.53 ± 0.29 | −83.26 ± 0.32 |
| mPuma | 2.62 | −11.65 | −124.60 ± 0.31 | −95.41 ± 0.28 | −84.97 ± 0.31 |
| mNoxaB | 14 | −10.67 | −113.63 ± 0.59 | −84.37 ± 0.51 | −71.03 ± 0.54 |
| mNoxaA | 36.9 | −10.09 | −111.86 ± 0.46 | −88.69 ± 0.39 | −83.16 ± 0.40 |
| mBax | 39.5 | −10.05 | −109.12 ± 0.44 | −82.81 ± 0.34 | −71.39 ± 0.36 |
| mHrk | 44.8 | −9.98 | −106.54 ± 0.44 | −80.12 ± 0.35 | −67.36 ± 0.38 |
| mBmf | 446 | −8.62 | −103.73 ± 0.40 | −74.39 ± 0.33 | −62.46 ± 0.36 |
| mBik | 658 | −8.39 | −95.75 ± 0.33 | −70.35 ± 0.30 | −56.26 ± 0.32 |
| PAP/BFE | ΔGvdw | ΔGele | ΔGpol | ΔGnon−pol | ΔGsol | ΔGgas | ΔGmmgbsa | ΔGbind | KD (nM) |
|---|---|---|---|---|---|---|---|---|---|
| mBak | −132.86 ± 0.29 | −233.23 ± 1.67 | 249.02 ± 1.49 | −18.83 ± 0.03 | 230.18 ± 1.48 | −366.10 ± 1.71 | −135.91 ± 0.39 | −12.05 | 1.33 |
| mBid | −140.93 ± 0.44 | −294.83 ± 2.30 | 331.54 ± 2.09 | −18.85 ± 0.05 | 369.33 ± 2.35 | −501.99 ± 2.69 | −132.66 ± 0.45 | −11.96 | 1.55 |
| mBim | −133.19 ± 0.30 | −389.53 ± 2.17 | 415.97 ± 2.02 | −18.51 ± 0.03 | 397.46 ± 2.01 | −522.73 ± 2.18 | −125.26 ± 0.34 | −11.77 | 2.16 |
| mPuma | −132.88 ± 0.24 | −391.12 ± 1.42 | 417.42 ± 1.27 | −18.02 ± 0.03 | 399.40 ± 1.25 | −524.01 ± 1.45 | −124.60 ± 0.31 | −11.65 | 2.62 |
| mNoxaB | −127.22 ± 0.42 | −142.90 ± 1.94 | 173.58 ± 1.77 | −17.09 ± 0.06 | 156.49 ± 1.74 | −270.13 ± 2.11 | −113.63 ± 0.59 | −10.67 | 14.0 |
| mNoxaA | −123.29 ± 0.40 | −148.49 ± 2.12 | 177.21 ± 2.12 | −17.29 ± 0.05 | 159.92 ± 2.09 | −271.78 ± 2.29 | −111.86 ± 0.46 | −10.09 | 36.9 |
| mBax | −135.79 ± 0.30 | −62.54 ± 2.25 | 106.89 ± 2.11 | −17.67 ± 0.04 | 70.17 ± 1.97 | −181.28 ± 2.10 | −109.12 ± 0.44 | −10.05 | 39.5 |
| mHrk | −120.76 ± 0.28 | −110.34 ± 3.28 | 140.73 ± 3.07 | −16.17 ± 0.04 | 124.56 ± 3.04 | −231.11 ± 3.37 | −106.54 ± 0.44 | −9.98 | 44.8 |
| mBmf | −129.10 ± 0.33 | −6.00 ± 1.57 | 48.56 ± 1.40 | −17.19 ± 0.05 | 31.37 ± 1.39 | −135.11 ± 1.61 | −103.73 ± 0.40 | −8.62 | 446 |
| mBik | −116.65 ± 0.33 | −119.83 ± 1.52 | 156.12 ± 1.35 | −15.39 ± 0.04 | 140.72 ± 1.35 | −236.48 ± 1.46 | −95.75 ± 0.33 | −8.39 | 658 |
| PAP/Pockets | Five Conserved Hydrophobic Residue Positions | ||||||
|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | G/A | D | P4 | P5 | |
| mBak | −4.53 ± 0.02 | −6.78 ± 0.02 | −3.91 ± 0.02 | −1.57 ± 0.03 | −2.47 ± 0.01 | −3.88 ± 0.02 | −2.86 ± 0.02 |
| mBid | −4.87 ± 0.02 | −6.54 ± 0.02 | −3.76 ± 0.02 | −1.65 ± 0.04 | −2.67 ± 0.01 | −3.75 ± 0.02 | −3.98 ± 0.02 |
| mBim | −5.65 ± 0.02 | −6.12 ± 0.02 | −3.71 ± 0.02 | −1.45 ± 0.03 | −2.09 ± 0.01 | −5.18 ± 0.02 | −3.63 ± 0.03 |
| mPuma | −4.29 ± 0.02 | −6.67 ± 0.02 | −4.31 ± 0.02 | −1.66 ± 0.02 | −3.08 ± 0.01 | −5.24 ± 0.02 | −2.46 ± 0.02 |
| mNoxaB | −0.29 ± 0.04 | −6.85 ± 0.03 | −3.33 ± 0.02 | −2.65 ± 0.04 | −2.73 ± 0.02 | −3.79 ± 0.02 | −4.50 ± 0.04 |
| mNoxaA | −6.19 ± 0.03 | −6.43 ± 0.02 | −4.53 ± 0.03 | −0.11 ± 0.01 | −1.92 ± 0.01 | −4.47 ± 0.02 | −5.89 ± 0.06 |
| mBax | −4.94 ± 0.03 | −5.92 ± 0.02 | −3.98 ± 0.02 | −1.36 ± 0.03 | −2.06 ± 0.01 | −4.36 ± 0.02 | −3.49 ± 0.02 |
| mHrk | −2.90 ± 0.02 | −5.97 ± 0.02 | −4.08 ± 0.02 | −1.10 ± 0.02 | −2.14 ± 0.01 | −5.17 ± 0.02 | −1.71 ± 0.01 |
| mBmf | −4.80 ± 0.03 | −5.81 ± 0.02 | −4.22 ± 0.02 | −0.34 ± 0.00 | −2.33 ± 0.01 | −5.72 ± 0.03 | −2.84 ± 0.02 |
| mBik | −4.26 ± 0.02 | −6.67 ± 0.02 | −3.52 ± 0.02 | −1.28 ± 0.02 | −2.43 ± 0.01 | −4.14 ± 0.02 | −3.33 ± 0.02 |
| PAPs/Residues | Residues Present in the Canonical Binding Groove of mMcl1 | ||||||
|---|---|---|---|---|---|---|---|
| R196 | V197 | V201 | H205 | F209 | M212 | V234 | |
| mBak | 0.10 ± 0.00 | −0.57 ± 0.01 | −1.05 ± 0.13 | −2.51 ± 0.31 | −0.97 ± 0.12 | −4.00 ± 0.25 | −2.54 ± 0.13 |
| mBid | −6.63 ± 0.05 | −0.54 ± 0.00 | −1.96 ± 0.14 | −2.63 ± 0.29 | −0.79 ± 0.09 | −3.82 ± 0.21 | −2.35 ± 0.21 |
| mBim | −0.19 ± 0.00 | −0.91 ± 0.01 | −2.20 ± 0.18 | −1.52 ± 0.31 | −0.79 ± 0.09 | −3.67 ± 0.21 | −3.04 ± 0.28 |
| mPuma | −4.53 ± 0.10 | −1.53 ± 0.01 | −1.48 ± 0.16 | −3.00 ± 0.30 | −0.83 ± 0.08 | −3.09 ± 0.21 | −3.81 ± 0.19 |
| mNoxaB | 0.07 ± 0.00 | −0.36 ± 0.00 | −0.80 ± 0.08 | −1.34 ± 0.15 | −0.92 ± 0.08 | −3.66 ± 0.22 | −3.48 ± 0.21 |
| mNoxaA | −3.47 ± 0.15 | −1.57 ± 0.01 | −2.08 ± 0.22 | −2.10 ± 0.29 | −0.85 ± 0.07 | −3.73 ± 0.21 | −3.36 ± 0.29 |
| mBax | −2.07 ± 0.02 | −2.13 ± 0.01 | −1.03 ± 0.13 | −1.69 ± 0.45 | −0.83 ± 0.09 | −3.15 ± 0.23 | −4.58 ± 0.23 |
| mHrk | 0.21 ± 0.00 | −0.19 ± 0.00 | −0.94 ± 0.09 | −1.62 ± 0.30 | −0.93 ± 0.08 | −3.88 ± 0.26 | −3.71 ± 0.20 |
| mBmf | −0.10 ± 0.00 | −1.24 ± 0.02 | −2.54 ± 0.21 | −3.14 ± 0.41 | −0.75 ± 0.09 | −3.60 ± 0.23 | −3.50 ± 0.30 |
| mBik | 0.10 ± 0.00 | −0.25 ± 0.00 | −0.85 ± 0.11 | −1.32 ± 0.27 | −0.98 ± 0.12 | −4.35 ± 0.22 | −2.55 ± 0.16 |
| N241 | G243 | R244 | T247 | L248 | F299 | F300 | |
| mBak | −2.01 ± 0.41 | −2.93 ± 0.3 | −4.99 ± 0.76 | −1.88 ± 0.18 | −1.44 ± 0.13 | −1.05 ± 0.37 | −1.44 ± 0.18 |
| mBid | −3.77 ± 0.34 | −3.66 ± 0.29 | −6.68 ± 0.69 | −2.28 ± 0.18 | −0.82 ± 0.09 | −1.52 ± 0.22 | −1.96 ± 0.22 |
| mBim | −1.36 ± 0.19 | −2.08 ± 0.24 | −4.86 ± 0.72 | −2.43 ± 0.20 | −1.05 ± 0.09 | −2.85 ± 0.36 | −3.42 ± 0.34 |
| mPuma | −1.22 ± 0.15 | −1.73 ± 0.16 | −4.88 ± 0.60 | −2.44 ± 0.15 | −1.07 ± 0.11 | −3.38 ± 0.26 | −2.86 ± 0.31 |
| mNoxaB | −3.36 ± 0.32 | −2.68 ± 0.24 | −8.18 ± 0.89 | −2.80 ± 0.23 | −0.67 ± 0.08 | −3.93 ± 0.42 | −2.74 ± 0.27 |
| mNoxaA | −0.82 ± 0.23 | −2.07 ± 0.23 | −2.69 ± 0.37 | −2.43 ± 0.23 | −1.19 ± 0.10 | −1.00 ± 0.22 | −1.79 ± 0.31 |
| mBax | −3.06 ± 0.84 | −3.19 ± 0.46 | −5.71 ± 0.83 | −1.89 ± 0.22 | −1.16 ± 0.12 | −0.17 ± 0.07 | −1.07 ± 0.20 |
| mHrk | −1.30 ± 0.16 | −2.61 ± 0.24 | −4.64 ± 0.54 | −2.19 ± 0.21 | −0.93 ± 0.10 | −3.35 ± 0.51 | −2.40 ± 0.25 |
| mBmf | −0.06 ± 0.04 | −1.45 ± 0.16 | −3.09 ± 0.53 | −2.33 ± 0.17 | −0.98 ± 0.10 | −2.22 ± 0.32 | −1.16 ± 0.14 |
| mBik | −1.66 ± 0.30 | −3.07 ± 0.30 | −5.44 ± 0.61 | −1.95 ± 0.23 | −0.86 ± 0.09 | −4.11 ± 0.35 | −1.24 ± 0.19 |
| Communities | Residues Involved |
|---|---|
| 1 | 147, 149, 151 |
| 2 | 175 |
| 3 | 177 |
| 4 | 302 |
| 5 | 304 |
| 6 | 307 |
| 7 | 308 |
| 8 | 93, 95 |
| 9 | 78 |
| 10 | 148, 150, 152 |
| 11 | 165–173, 253, 255–269, 271 |
| 12 | 174, 176, 178–193, 195, 293–294, 297, 299, 301, 303, 305–306 |
| 13 | 194, 196–206, 208–209, 237, 239, 241–252, 254,2 91, 295–296, 298, 300, 78–80, 82 |
| 14 | 75, 77, 81, 83–92, 94, 96 |
| 15 | 207, 210–216, 226, 228, 229–236, 238 |
| 16 | 153–164, 240, 270, 272–290, 292 |
| 17 | 217–225, 227 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marimuthu, P.; Razzokov, J.; Singaravelu, K.; Bogaerts, A. Predicted Hotspot Residues Involved in Allosteric Signal Transmission in Pro-Apoptotic Peptide—Mcl1 Complexes. Biomolecules 2020, 10, 1114. https://doi.org/10.3390/biom10081114
Marimuthu P, Razzokov J, Singaravelu K, Bogaerts A. Predicted Hotspot Residues Involved in Allosteric Signal Transmission in Pro-Apoptotic Peptide—Mcl1 Complexes. Biomolecules. 2020; 10(8):1114. https://doi.org/10.3390/biom10081114
Chicago/Turabian StyleMarimuthu, Parthiban, Jamoliddin Razzokov, Kalaimathy Singaravelu, and Annemie Bogaerts. 2020. "Predicted Hotspot Residues Involved in Allosteric Signal Transmission in Pro-Apoptotic Peptide—Mcl1 Complexes" Biomolecules 10, no. 8: 1114. https://doi.org/10.3390/biom10081114
APA StyleMarimuthu, P., Razzokov, J., Singaravelu, K., & Bogaerts, A. (2020). Predicted Hotspot Residues Involved in Allosteric Signal Transmission in Pro-Apoptotic Peptide—Mcl1 Complexes. Biomolecules, 10(8), 1114. https://doi.org/10.3390/biom10081114