Colorectal Cancer Prediction Based on Weighted Gene Co-Expression Network Analysis and Variational Auto-Encoder


Abstract
:1. Introduction
2. Materials and Methods
2.1. CRC Microarray Datasets
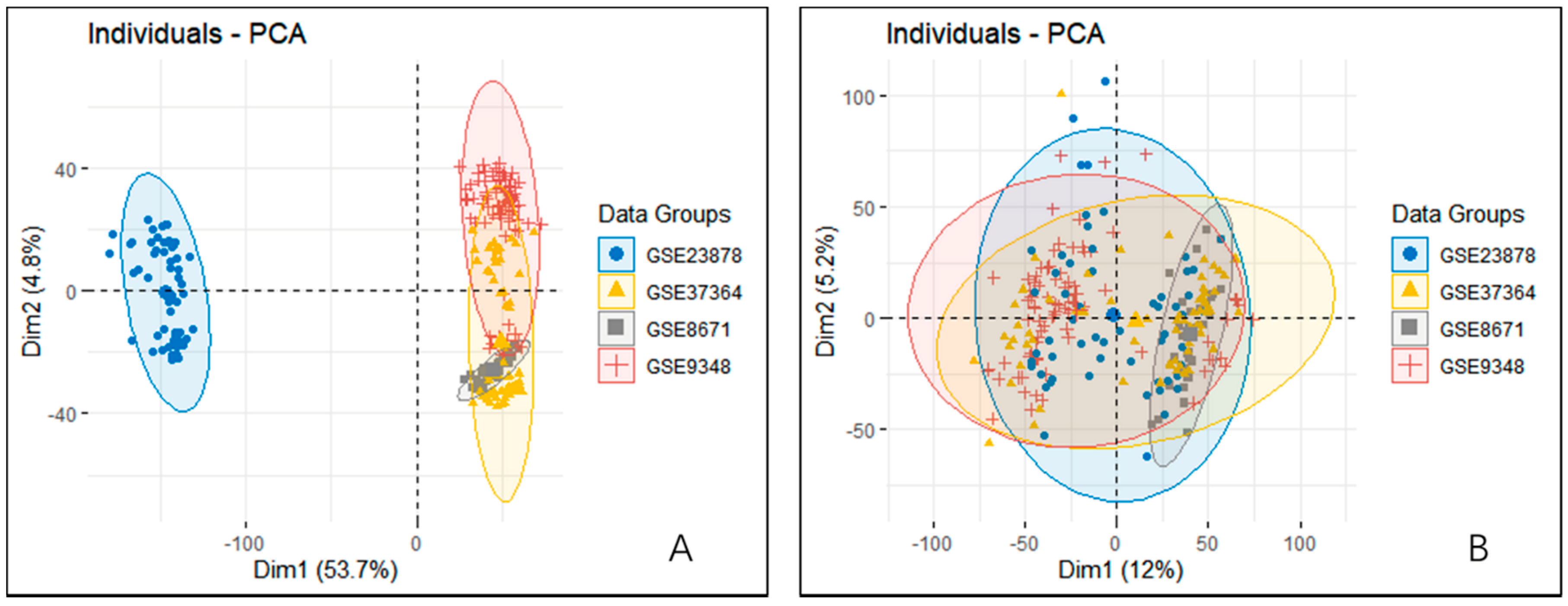
2.2. Datasets Preprocessing
2.3. Screening of Differentially Expressed Genes
2.4. Construction of Co-Expression Network and Identification of Key Modules
2.5. Mining Hub Genes from the Key Module
2.6. Dimension Reduction with VAE
2.7. SVM
3. Results and Discussion
3.1. Extraction of Differential Genes
3.2. Extracting Hub Gene by Weighted Correlation Network Analysis
3.2.1. Soft Threshold Screening
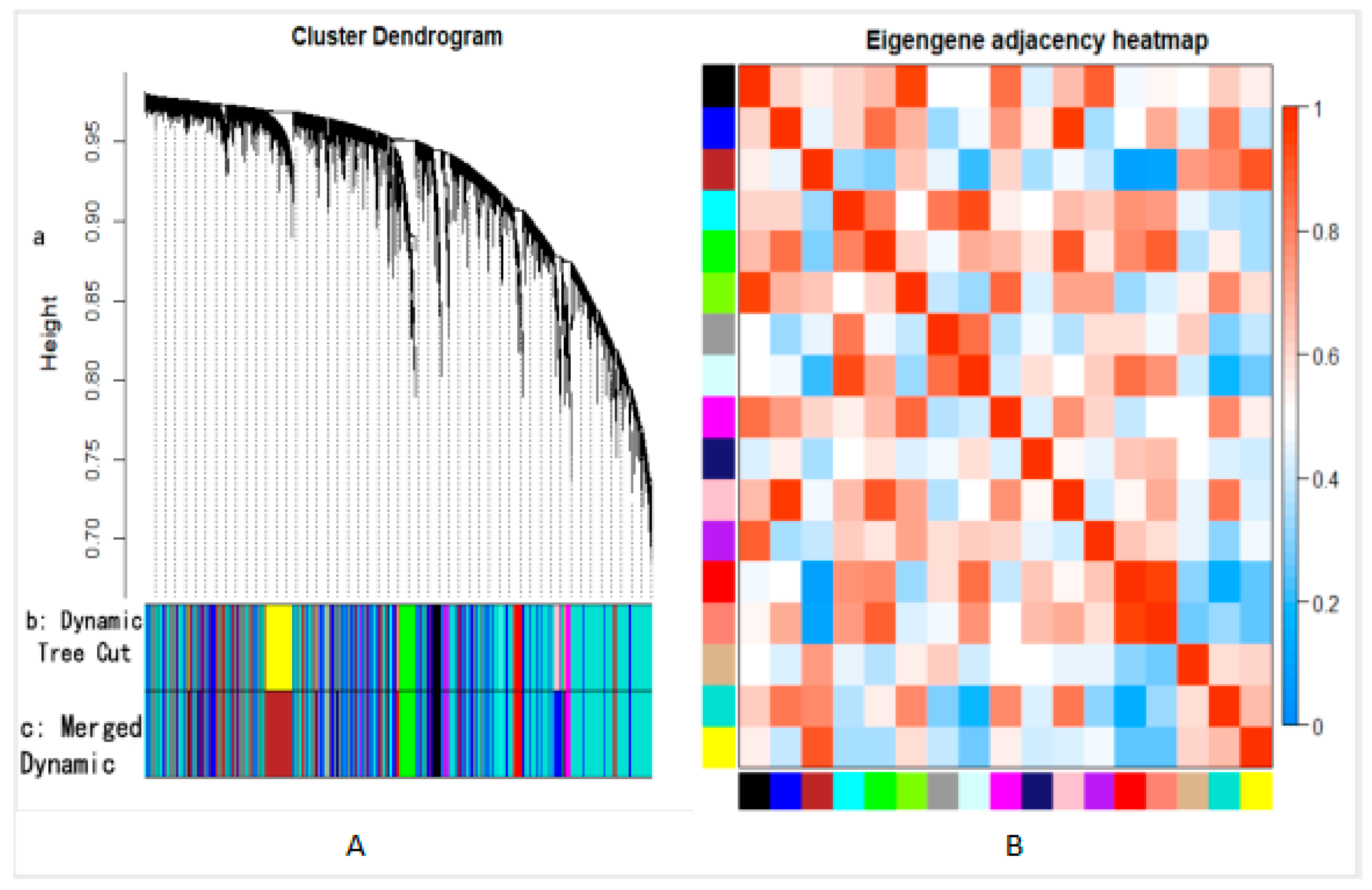
3.2.2. Gene Module Construction
3.2.3. Hub Gene Identification
3.3. Realization of VAE
3.4. Analysis Results from the Model of Cancer Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.F.J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, L.; Tian, H.; Chen, Z.; Chen, H.; Xu, J.; Fang, J.Y. Systematic evaluation of supervised classifiers for fecal microbiota-based prediction of colorectal cancer. Oncotarget 2017, 8, 9546–9556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.L.; Nevado, A.; Carro, B. Detection of early stages of Alzheimer’s disease based on MEG activity with a randomized convolutional neural network. Artif. Intell. Med. 2020, 107, 101924. [Google Scholar]
- Zhao, D.D.; Liu, H.; Zheng, Y.J.; He, Y.L.; Lu, D.J.; Chen, L. A reliable method for colorectal cancer prediction based on feature selection and support vector machine. Med. Biol. Eng. Comput. 2019, 577, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Agesen, T.H.; Sevvn, A.; Lind, G.E.; Nesbakken, A.; Skotheim, R.L.; Lothe, R.A. ColoGuideEx: A robust gene classifier specific for stage II colorectal cancer prognosis. Gut 2012, 61, 1560–1567. [Google Scholar] [CrossRef]
- Gabere, M.N.; Hussein, N.A.; Aziz, M.A. Filtered selection coupled with support vector machines generate a functionally relevant prediction model for colorectal cancer. Oncotargets Ther. 2016, 9, 3313–3325. [Google Scholar]
- Cubiella, J.; Vega, P.; Salve, M.; Ondina, M.D.; Alves, M.T.; Quintero, E.; Victoria, Á.S.; Fernando, F.B.; Boadas, J.; Campo, R. Development and external validation of a faecal immunochemical test-based prediction model for colorectal cancer detection in symptomatic patients. BMC Med. 2016, 14, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Karabulut, E.M.; Ibrikci, T. Discriminative deep belief networks for microarray based cancer classification. Biomed. Res. 2017, 28, 1016–1024. [Google Scholar]
- Yong, F.L.; Law, C.W.; Wang, C.W. Potentiality of a triple microRNA classifier: miR-193a-3p, miR-23a and miR-338-5p for early detection of colorectal cancer. BMC Cancer 2013, 13, 280. [Google Scholar] [CrossRef] [Green Version]
- Bärlund, M.; Monni, O.; Kononen, J.; Cornelison, R.; Torhorst, J.; Sauter, G.; Kallioniemi, O.P.; Kallioniemi, A. Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer Res. 2000, 60, 5340–5344. [Google Scholar]
- Carlson, M.R.; Zhang, B.; Fang, Z.; Mischel, P.S.; Horvath, S.; Nelson, S.F. Gene connectivity, function, and sequence conservation: Predictions from modular yeast co-expression networks. BMC Genom. 2006, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Zhao, J.; Fan, X.; Kang, Z. Weighted gene co-expression network analysis in identification of metastasis-related genes of lung squamous cell carcinoma based on the Cancer Genome Atlas database. J. Thorac. Dis. 2017, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Zeng, J.; Shi, N.; Chen, L.; Wang, L. Application of Weighted Gene co-expression Network Analysis to Explore the Potential Diagnostic Biomarkers for Colorectal Cancer. Mol. Med. Rep. 2020, 21, 2533–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, M.; Müller, F.-J.; Zenke, M.; Schuppert, A. Principal components analysis and the reported low intrinsic dimensionality of gene expression microarray data. Sci. Rep. 2016, 6, 25696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta, E.B.; Duval, B.; Hao, J.K. A hybrid LDA and genetic algorithm for gene selection and classification of microarray data. Neurocomputing 2010, 73, 2375–2383. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yao, H.; Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing 2016, 184, 232–242. [Google Scholar] [CrossRef]
- Shin, H.C.; Orton, M.R.; Collins, D.J.; Doran, S.J.; Leach, M.O. Stacked autoencoders for unsupervised feature learning and multiple organ detection in a pilot study using 4D patient data. IEEE Trans. Pattern Anal. Mach. Intell. 2012, 35, 1930–1943. [Google Scholar] [CrossRef]
- Ng, A. Sparse autoencoder. CS294A Lect. Notes 2011, 72, 1–19. [Google Scholar]
- Vincent, P.; Larochelle, H.; Bengio, Y.; Manzagol, P.A. Extracting and Composing Robust Features with Denoising Autoencoders. In Proceedings of the 25th International Conference on Machine Learning, Association for Computing Machinery, New York, NY, USA, 2014. [Google Scholar]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. In Proceedings of the International Conference on Learning Representations, Banff, AB, Canada, 14–16 April 2014. [Google Scholar]
- Chatrian, A.; Sirinukunwattana, K.; Verrill, C.; Rittscher, J. Towards the Identification of Histology Based Subtypes in Prostate Cancer. In Proceedings of the International Symposium on Biomedical Imaging, Venice, Italy, 24–27 April 2019. [Google Scholar]
- Wang, Z.X.; Wang, Y.D. Extracting a biologically latent space of lung cancer epigenetics variational autoencoders. BMC Bioinform. 2019, 20, 568. [Google Scholar] [CrossRef]
- Sabates-Bellver, J.; Van der Flier, L.G.; de Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007, 5, 1263–1275. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Downey, T.; Eu, K.W.; Koh, P.K.; Cheah, P.Y. A ‘metastasis-prone’signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin. Exp. Metastasis 2010, 27, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Ahmed, M.; Hussain, A.; Abubaker, J.; Al-Sanea, N.; AbdulJabbar, A.; Ashari, L.H.; Alhomoud, S.; Al-Dayel, F.; Jehan, Z.; et al. Genome-wide expression analysis of Middle Eastern colorectal cancer reveals FOXM1 as a novel target for cancer therapy. Am. J. Pathol. 2011, 178, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valcz, G.; Patai, Á.V.; Kalmár, A.; Péterfia, B.; Fűri, I.; Wichmann, B.; Műzes, G.; Sipos, F.; Krenács, T.; Mihály, E. Myofibroblast-derived SFRP1 as potential inhibitor of colorectal carcinoma field effect. PloS ONE 2014, 9, E106143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Churchill, G.A. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003, 4, 210. [Google Scholar] [CrossRef] [Green Version]
- Bevilacqua, V.; Pannarale, P.; Abbrescia, M.; Cava, C.; Paradiso, A.; Tommasi, S. Comparison of data-merging methods with SVM attribute selection and classification in breast cancer gene expression. BMC Bioinform. 2012, 13, S9. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Schumacher, M.; Scherer, A.; Sanoudou, D.; Megherbi, D.; Davison, T.; Shi, T.; Tong, W.; Shi, L.; Hong, H.; et al. A comparison of batch effect removal methods for enhancement of prediction performance using MAQC-II microarray gene expression data. Pharm. J. 2010, 10, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Alter, O.; Brown, P.O.; Botstein, D. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. USA 2000, 97, 10101–10106. [Google Scholar] [CrossRef] [Green Version]
- Benito, M.; Parker, J.; Du, Q.; Wu, J.; Xiang, D.; Perou, C.M.; Marron, J.S. Adjustment of systematic microarray data biases. Bioinformatics 2004, 20, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.K.; Qu, P.; Epstein, J.; Buros, A.; Rosenthal, A.; Crowley, J.; Morgan, G.; Barlogie, B. Removing batch effects from purified plasma cell gene expression microarrays with modified ComBat. BMC Bioinform. 2015, 16, 63. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, D.; Lu, M.; Xu, J.; Austin, C.; Caskey, C.T.; Rushmore, T. Monitoring expression of genes involved in drug metabolism and toxicology using DNA microarrays. Physiol. Genom. 2001, 5, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Long, A.D. A Bayesian framework for the analysis of microarray expression data: Regularized t-test and statistical inferences of gene changes. Bioinformatics 2001, 17, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, E47. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2007, 24, 719–720. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 1. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Lou, Y.; Tian, G.Y.; Song, Y.; Liu, Y.L.; Chen, Y.D.; Shi, J.P.; Yang, J. Characterization of transcriptional modules related to fibrosing-NAFLD progression. Sci. Rep. 2017, 7, 4748. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Pan, J.; Xin, Y.; Mi, X.; Wang, J.; Gao, Q.; Luo, H. Gene Expression Analysis Reveals Novel Gene Signatures Between Young and Old Adults in Human Prefrontal Cortex. Front. Aging Neurosci. 2018, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Foody, G.M.; Mathur, A. Toward intelligent training of supervised image classifications: Directing training data acquisition for SVM classification. Remote Sens. Environ. 2004, 1, 107–117. [Google Scholar] [CrossRef]
- Meeh, C.L.; Croshaw, R.; Crimm, H.; Miller, S.K.; Oroian, D.; Kowli, S.; Zhu, J.; Carver, W.; Wu, W.; Pena, E.A.; et al. A Gene Expression Classifier of Node-Positive Colorectal Cancer. Neoplasia 2009, 11, 1074–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, K. Determination of the coefficient of correlation. Science 1909, 30, 23–25. [Google Scholar] [CrossRef]
- Nagaraj, S.H.; Reverter, A. A Boolean-based systems biology approach to predict novel genes associated with cancer: Application to colorectal cancer. BMC Syst. Biol. 2011, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Helms, T.L.; Feng, N.; Chang, Q.E.; Tian, F.; Wu, J.Y.; Toniatti, C.; Heffernan, T.P.; Powis, G.; Kwong, L.N.; et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016, 26, 39595–39608. [Google Scholar] [CrossRef] [Green Version]
- Kurita, K.; Maeda, M.; Mansour, M.A.; Kokuryo, T.; Uehara, K.; Yokoyama, Y.; Nagino, M.; Hamaguchi, M.; Senga, T. TRIP13 is expressed in colorectal cancer and promotes cancer cell invasion. Oncol. Lett. 2016, 12, 5240–5246. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, J.; Sun, J.; Cui, Z.; Wu, H. RNA interference-mediated silencing of eukaryotic translation initiation factor 3, subunit B (EIF3B) gene expression inhibits proliferation of colon cancer cells. World J. Surg. Oncol. 2012, 10, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Alimperti, S.; Andreadis, S.T. CDH2 and CDH11 act as regulators of stem cell fate decisions. Stem Cell Res. 2015, 14, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Kumara, H.S.; Bellini, G.A.; Caballero, O.L.; Herath, S.A.; Su, T.; Ahmed, A.; Njoh, L.; Ckkic, V.; Whelan, R.L. P-Cadherin (CDH3) is overexpressed in colorectal tumors and has potential as a serum marker for colorectal cancer monitoring. Oncoscience 2017, 4, 139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Du, Y.; Wang, Z.; Lou, R.; Wu, J.; Feng, J. Integrated Analysis of Oncogenic Networks in Colorectal Cancer Identifies GUCA2A as a Molecular Marker. Biochem. Res. Int. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Color | Tan | Brown | Turquoise | Blue | Green | Purple |
|---|---|---|---|---|---|---|
| Number | 85 | 2799 | 6377 | 2636 | 400 | 153 |
| Color | Black | Magenta | Midnight Blue | Red | Cyan | Grey60 |
| Number | 330 | 175 | 60 | 318 | 118 | 36 |
| MEtan | MEbrown | MEturquoise | MEblue | MEgreen | MEpurple | |
|---|---|---|---|---|---|---|
| PCC | −0.1285 | −0.3052 | −0.9251 | −0.7075 | −0.2017 | 0.3457 |
| p-value | 0.0920 | 0.0000 | 0.0000 | 0.0000 | 0.0078 | 0.0000 |
| MEblack | MEmagenta | MEblue | MEred | MEcyan | MEgrey60 | |
| PCC | −0.4263 | −0.5753 | 0.0609 | 0.5127 | 0.2944 | 0.3082 |
| p-value | 0.0000 | 0.0000 | 0.4260 | 0.0000 | 0.0000 | 0.0000 |
| GENE NAME | logFC | adj.P.Val | GS | MM.Turquoise | K.in |
|---|---|---|---|---|---|
| CDK4 | 1.3042 | 2.40 × 10−39 | 0.8128 | −0.9076 | 933.8071 |
| CDH3 | 6.3918 | 2.92 × 10−80 | 0.9417 | −0.9206 | 922.1908 |
| DKC1 | 1.3517 | 3.34 × 10−41 | 0.8226 | −0.9055 | 918.2304 |
| UBE2S | 1.9085 | 7.08 × 10−40 | 0.8118 | −0.8997 | 906.0183 |
| GUCA2B | −6.4444 | 6.12 × 10−53 | 0.8723 | 0.9075 | 895.3242 |
| UBE2C | 2.1435 | 9.16 × 10−52 | 0.8706 | −0.9122 | 895.1551 |
| EIF3B | 1.3173 | 1.02 × 10−43 | 0.8368 | −0.8970 | 894.6524 |
| TRIP13 | 1.9410 | 2.07 × 10−46 | 0.8474 | −0.8905 | 890.4061 |
| GUCA2A | −5.3201 | 2.29 × 10−50 | 0.8628 | 0.8628 | 887.2683 |
| GTF3A | 1.4126 | 5.21 × 10−39 | 0.8085 | −0.8173 | 883.3120 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ai, D.; Wang, Y.; Li, X.; Pan, H. Colorectal Cancer Prediction Based on Weighted Gene Co-Expression Network Analysis and Variational Auto-Encoder. Biomolecules 2020, 10, 1207. https://doi.org/10.3390/biom10091207
Ai D, Wang Y, Li X, Pan H. Colorectal Cancer Prediction Based on Weighted Gene Co-Expression Network Analysis and Variational Auto-Encoder. Biomolecules. 2020; 10(9):1207. https://doi.org/10.3390/biom10091207
Chicago/Turabian StyleAi, Dongmei, Yuduo Wang, Xiaoxin Li, and Hongfei Pan. 2020. "Colorectal Cancer Prediction Based on Weighted Gene Co-Expression Network Analysis and Variational Auto-Encoder" Biomolecules 10, no. 9: 1207. https://doi.org/10.3390/biom10091207
APA StyleAi, D., Wang, Y., Li, X., & Pan, H. (2020). Colorectal Cancer Prediction Based on Weighted Gene Co-Expression Network Analysis and Variational Auto-Encoder. Biomolecules, 10(9), 1207. https://doi.org/10.3390/biom10091207