
Interplay between Epigenetics and Cellular Metabolism in Colorectal Cancer


Abstract
:1. Introduction
2. Epigenetics in CRC
2.1. DNA Methylation and Hydroxymethylation in CRC
2.1.1. DNA Methylation in CRC
2.1.2. DNA Hydroxymethylation in CRC
2.2. Histone Post-Translational Modification
2.3. Non-Coding RNAs in CRC
2.3.1. MicroRNAs in CRC
2.3.2. LncRNAs in CRC
3. Cellular Metabolism in CRC
3.1. Aerobic Glycolysis in CRC
3.2. Glutamine Metabolism in CRC
3.3. Biosynthetic Metabolism in CRC
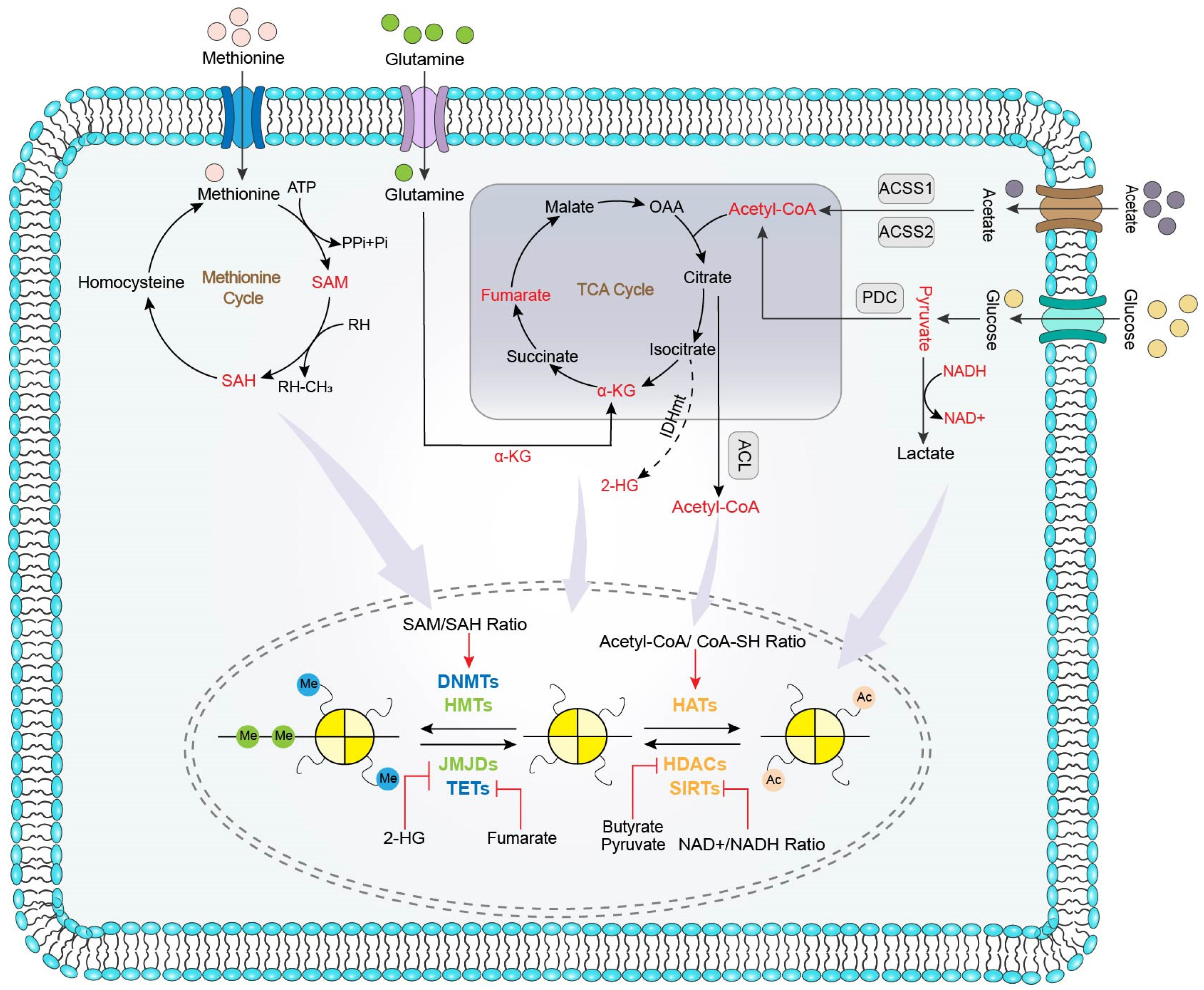
4. Metabolism Influences Epigenetic in CRC
4.1. Metabolites Influences Epigenetic Process
4.1.1. Methyl Donor Regulates Epigenetics in CRC
4.1.2. Acetyl Donor Regulates Epigenetics in CRC
4.2. Metabolites Influences the Activity of Epigenetic Enzyme
4.2.1. Metabolites Affecting DNMTs and HATs
4.2.2. Metabolites Affecting JMJDs and TETs
4.2.3. Metabolites Affecting HATs
4.2.4. Metabolites Affecting HDACs and SIRTs
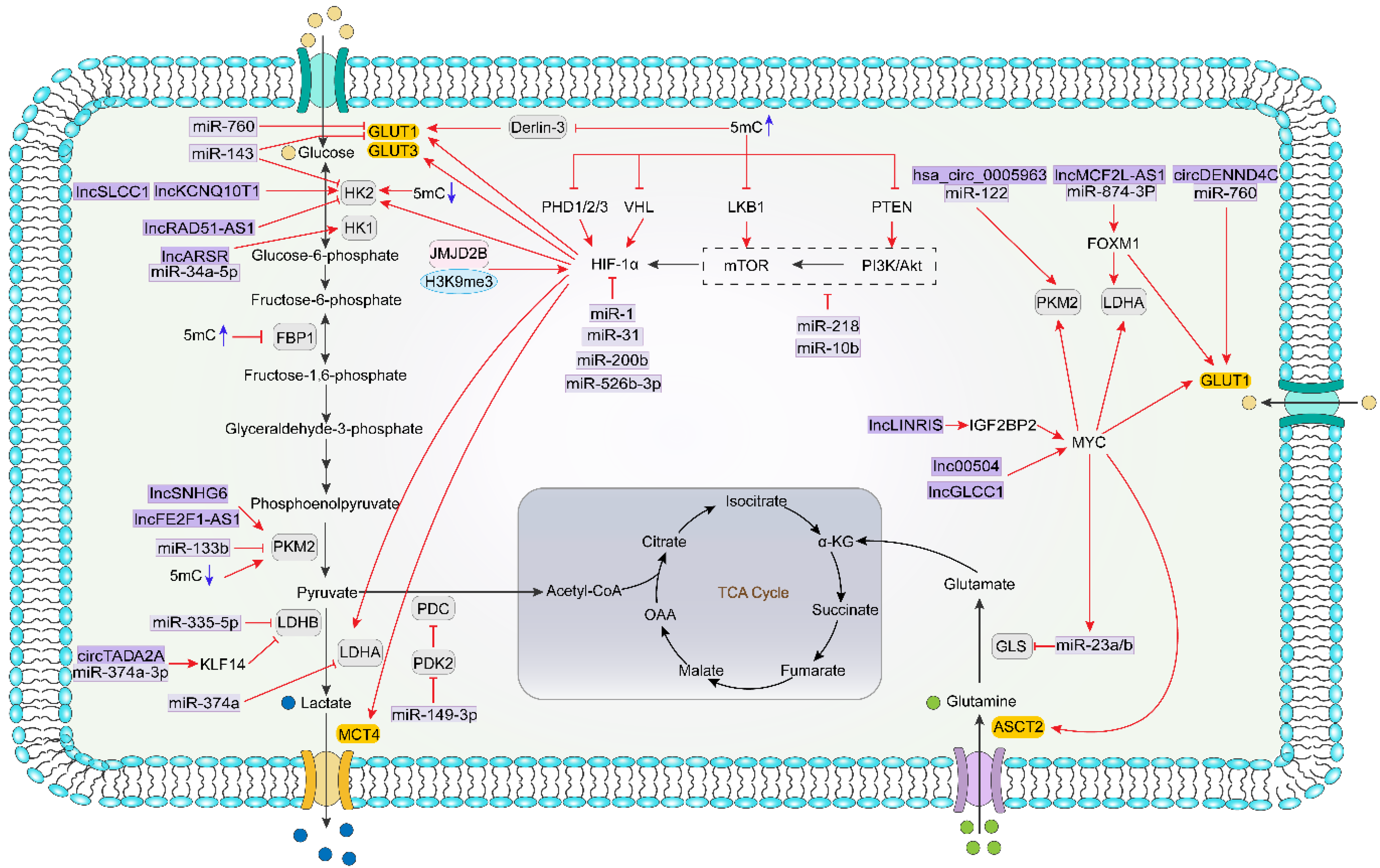
5. Epigenetics Influence Metabolism in CRC
5.1. Epigenetics Direct Regulation of Metabolism-Related Genes
5.2. Epigenetics Indirectly Regulate Cellular Metabolism via Modulating Metabolism-Related Transcription Factors and Signaling Pathways
6. Epigenetic-Metabolic Crosstalk as a Therapeutic Target of CRC
6.1. Metabolism Inhibitors
6.1.1. Glycolysis Inhibitors
6.1.2. Glutaminase Inhibitors
6.1.3. SAM Cycle Inhibitors
6.2. Epigenetic Inhibitors
6.2.1. DNMT Inhibitors
6.2.2. HDAC Inhibitors
6.2.3. SIRT Activators and Inhibitors
6.2.4. MiRNA Modulators
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Purushothaman, V.L.; Cuomo, R.E.; Garland, C.F.; Mackey, T.K. Could age increase the strength of inverse association between ultraviolet B exposure and colorectal cancer? BMC Public Health 2021, 21, 1238. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Jair, K.-W.; Bachman, K.E.; Suzuki, H.; Ting, A.H.; Rhee, I.; Yen, R.-W.C.; Baylin, S.B.; Schuebel, K.E. De novo CpG island methylation in human cancer cells. Cancer Res. 2006, 66, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- Qin, J.; Wen, B.; Liang, Y.; Yu, W.; Li, H. Histone Modifications and their Role in Colorectal Cancer (Review). Pathol. Oncol. Res. 2020, 26, 2023–2033. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.; Tong, C.W.; Wu, M.; Cho, W.C. MicroRNAs in the prognosis and therapy of colorectal cancer: From bench to bedside. World J. Gastroenterol. 2018, 24, 2949–2973. [Google Scholar] [CrossRef] [PubMed]
- Dastmalchi, N.; Safaralizadeh, R.; Nargesi, M.M. LncRNAs: Potential Novel Prognostic and Diagnostic Biomarkers in Colorectal Cancer. Curr. Med. Chem. 2020, 27, 5067–5077. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, M.; Aguilar-Medina, M.; Lizárraga-Verdugo, E.; Avendaño-Félix, M.; Silva-Benítez, E.; López-Camarillo, C.; Ramos-Payán, R. LncRNAs as Regulators of Autophagy and Drug Resistance in Colorectal Cancer. Front. Oncol. 2019, 9, 1008. [Google Scholar] [CrossRef]
- Dong, Z.; Pu, L.; Cui, H. Mitoepigenetics and Its Emerging Roles in Cancer. Front. Cell Dev. Biol. 2020, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.; Hou, J.; Cui, H. Serine-glycine-one-carbon metabolism: Vulnerabilities in MYCN-amplified neuroblastoma. Oncogenesis 2020, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2021, 1–43. [Google Scholar] [CrossRef]
- Aspeslagh, S.; Morel, D.; Soria, J.C.; Postel-Vinay, S. Epigenetic modifiers as new immunomodulatory therapies in solid tumours. Ann. Oncol. 2018, 29, 812–824. [Google Scholar] [CrossRef]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2019, 57, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Dong, Z.; Ke, X.; Hou, J.; Zhao, E.; Zhang, K.; Wang, F.; Yang, L.; Xiang, Z.; Cui, H. The roles of sirtuins family in cell metabolism during tumor development. Semin. Cancer Biol. 2019, 57, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Cui, H. Chapter 11—Sirtuins and cellular metabolism in cancers. In Sirtuin Biology in Cancer and Metabolic Disease; Maiese, K., Ed.; Academic Press: London, UK, 2021; pp. 195–217. [Google Scholar] [CrossRef]
- Margot, J.B.; Cardoso, M.C.; Leonhardt, H. Mammalian DNA methyltransferases show different subnuclear distributions. J. Cell. Biochem. 2001, 83, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef]
- Peng, X.; Qing, Z. CpG island methylation of tumor suppressor genes in colorectal cancer. Chin. J. Gen. Surg. 2012, 21, 1279–1282. [Google Scholar]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrami, A.; Hassanian, S.M.; Khazaei, M.; Gharib, M.; Rahmani, M.; Fiuji, H.; Jazayeri, M.H.; Moetamani-Ahmadi, M.; Ferns, G.A.; Avan, A. The 9p21 locus as a potential therapeutic target and prognostic marker in colorectal cancer. Pharmacogenomics 2018, 19, 463–474. [Google Scholar] [CrossRef]
- Cavestro, G.M.; Mannucci, A.; Zuppardo, R.A.; Di Leo, M.; Stoffel, E.; Tonon, G. Early onset sporadic colorectal cancer: Worrisome trends and oncogenic features. Dig. Liver Dis. 2018, 50, 521–532. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst. 2008, 100, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- Lam, K.; Pan, K.; Linnekamp, J.F.; Medema, J.P.; Kandimalla, R. DNA methylation based biomarkers in colorectal cancer: A systematic review. Biochim. Biophys. Acta 2016, 1866, 106–120. [Google Scholar] [CrossRef]
- Dong, Z.; Dai, L.; Zhang, Y.; Fang, C.; Shi, G.; Chen, Y.; Li, J.; Wang, Q.; Fu, J.; Yu, Y.; et al. Hypomethylation of GDNF family receptor alpha 1 promotes epithelial-mesenchymal transition and predicts metastasis of colorectal cancer. PLoS Genet. 2020, 16, e1009159. [Google Scholar] [CrossRef]
- Jeschke, J.; Collignon, E.; Fuks, F. Portraits of TET-mediated DNA hydroxymethylation in cancer. Curr. Opin. Genet. Dev. 2016, 36, 16–26. [Google Scholar] [CrossRef]
- Chapman, C.G.; Mariani, C.J.; Wu, F.; Meckel, K.; Butun, F.; Chuang, A.; Madzo, J.; Bissonnette, M.B.; Kwon, J.H.; Godley, L.A. TET-catalyzed 5-hydroxymethylcytosine regulates gene expression in differentiating colonocytes and colon cancer. Sci. Rep. 2015, 5, 17568. [Google Scholar] [CrossRef] [Green Version]
- Zhong, A.; Tian, Y.; Zhang, H.; Lai, M. DNA hydroxymethylation of colorectal primary carcinoma and its association with survival. J. Surg. Oncol. 2018, 117, 1029–1037. [Google Scholar] [CrossRef]
- Li, M.; Gao, F.; Xia, Y.; Tang, Y.; Zhao, W.; Jin, C.; Luo, H.; Wang, J.; Li, Q.; Wang, Y. Filtrating colorectal cancer associated genes by integrated analyses of global DNA methylation and hydroxymethylation in cancer and normal tissue. Sci. Rep. 2016, 6, 31826. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Lewis, S.; Stark, R.; Carroll, T.; Dunning, M.J.; Bachman, M.; Ito, Y.; Stojic, L.; Halim, S.; Vowler, S.L.; Lynch, A.G.; et al. 5-hydroxymethylcytosine marks promoters in colon that resist DNA hypermethylation in cancer. Genome Biol. 2015, 16, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerecke, C.; Schumacher, F.; Edlich, A.; Wetzel, A.; Yealland, G.; Neubert, L.K.; Scholtka, B.; Homann, T.; Kleuser, B. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget 2018, 9, 32822–32840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Mo, M.; Zhong, Y.; Yang, Q.; Zhang, J.; Ye, X.; Zhang, L.; Cai, C. The Anticancer Role of Omega-3 Polyunsaturated Fatty Acids was Closely Associated with the Increase in Genomic DNA Hydroxymethylation. Anti-Cancer Agents Med. Chem. 2019, 19, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Hebbes, T.R.; Thorne, A.W.; Crane-Robinson, C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988, 7, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Karczmarski, J.; Rubel, T.; Paziewska, A.; Mikula, M.; Bujko, M.; Kober, P.; Dadlez, M.; Ostrowski, J. Histone H3 lysine 27 acetylation is altered in colon cancer. Clin. Proteom. 2014, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashktorab, H.; Belgrave, K.; Hosseinkhah, F.; Brim, H.; Nouraie, M.; Takkikto, M.; Hewitt, S.; Lee, E.L.; Dashwood, R.H.; Smoot, D. Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Dig. Dis. Sci. 2009, 54, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Benard, A.; Goossens-Beumer, I.J.; van Hoesel, A.Q.; de Graaf, W.; Horati, H.; Putter, H.; Zeestraten, E.C.M.; van de Velde, C.J.H.; Kuppen, P.J.K. Histone trimethylation at H3K4, H3K9 and H4K20 correlates with patient survival and tumor recurrence in early-stage colon cancer. BMC Cancer 2014, 14, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valencia, A.M.; Kadoch, C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat. Cell Biol. 2019, 21, 152–161. [Google Scholar] [CrossRef]
- Yin, C.; Ke, X.; Zhang, R.; Hou, J.; Dong, Z.; Wang, F.; Zhang, K.; Zhong, X.; Yang, L.; Cui, H. G9a promotes cell proliferation and suppresses autophagy in gastric cancer by directly activating mTOR. FASEB J. 2019, 33, 14036–14050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Zou, J.; Li, J.; Pang, Y.; Liu, Y.; Deng, C.; Chen, F.; Cui, H. MYST1/KAT8 contributes to tumor progression by activating EGFR signaling in glioblastoma cells. Cancer Med. 2019, 8, 7793–7808. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Yang, J.; Li, L.; Tan, L.; Shi, P.; Zhang, J.; Zhong, X.; Ge, L.; Wu, Z.; Cui, H. FOXO3a-SIRT6 axis suppresses aerobic glycolysis in melanoma. Int. J. Oncol. 2020, 56, 728–742. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Zhong, X.; Lei, Q.; Chen, F.; Cui, H. Transcriptional activation of SIRT6 via FKHRL1/FOXO3a inhibits the Warburg effect in glioblastoma cells. Cell. Signal. 2019, 60, 100–113. [Google Scholar] [CrossRef]
- Yang, L.; Lei, Q.; Li, L.; Yang, J.; Dong, Z.; Cui, H. Silencing or inhibition of H3K79 methyltransferase DOT1L induces cell cycle arrest by epigenetically modulating c-Myc expression in colorectal cancer. Clin. Epigenet. 2019, 11, 199. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Kou, L.; Yu, L.; Bai, C.; Li, M.; Mo, P.; Li, W.; Yu, C. Histone Demethylase JMJD2D Interacts With β-Catenin to Induce Transcription and Activate Colorectal Cancer Cell Proliferation and Tumor Growth in Mice. Gastroenterology 2019, 156, 1112–1126. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.N.; Zhi, Z.; Chen, L.Y.; Zhou, Q.; Li, X.M.; Gan, W.J.; Chen, S.; Yang, M.; Liu, Y.; Shen, T.; et al. SIRT1 suppresses colorectal cancer metastasis by transcriptional repression of miR-15b-5p. Cancer Lett. 2017, 409, 104–115. [Google Scholar] [CrossRef]
- Tampakis, A.; Tampaki, E.C.; Nebiker, C.A.; Kouraklis, G. Histone deacetylase inhibitors and colorectal cancer: What is new? Anticancer Agents Med. Chem. 2014, 14, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Balacescu, O.; Sur, D.; Cainap, C.; Visan, S.; Cruceriu, D.; Manzat-Saplacan, R.; Muresan, M.-S.; Balacescu, L.; Lisencu, C.; Irimie, A. The Impact of miRNA in Colorectal Cancer Progression and Its Liver Metastases. Int. J. Mol. Sci. 2018, 19, 3711. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Lei, W.; Fu, J.-C.; Zhang, L.; Li, J.-H.; Xiong, J.-P. Targeting miR-21 enhances the sensitivity of human colon cancer HT-29 cells to chemoradiotherapy in vitro. Biochem. Biophys. Res. Commun. 2014, 443, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Akçakaya, P.; Ekelund, S.; Kolosenko, I.; Caramuta, S.; Ozata, D.M.; Xie, H.; Lindforss, U.; Olivecrona, H.; Lui, W.-O. miR-185 and miR-133b deregulation is associated with overall survival and metastasis in colorectal cancer. Int. J. Oncol. 2011, 39, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Chen, D.; Li, X.; Yang, K.; Wang, H.; Wu, W. miR-133b regulates the MET proto-oncogene and inhibits the growth of colorectal cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 10, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Gomes, S.E.; Simões, A.E.S.; Pereira, D.M.; Castro, R.E.; Rodrigues, C.M.P.; Borralho, P.M. miR-143 or miR-145 overexpression increases cetuximab-mediated antibody-dependent cellular cytotoxicity in human colon cancer cells. Oncotarget 2016, 7, 9368–9387. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cho, K.B.; Li, Y.; Tao, G.; Xie, Z.; Guo, B. Long Noncoding RNA (lncRNA)-Mediated Competing Endogenous RNA Networks Provide Novel Potential Biomarkers and Therapeutic Targets for Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 5758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Weng, W.; Zhang, Q.; Wu, Y.; Ni, S.; Tan, C.; Xu, M.; Sun, H.; Liu, C.; Wei, P.; et al. The lncRNA NEAT1 activates Wnt/β-catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J. Hematol. Oncol. 2018, 11, 113. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.F.; Yin, Q.F.; Chen, T.; Zhang, Y.; Zhang, X.O.; Wu, Z.; Zhang, S.; Wang, H.B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014, 24, 513–531. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.; Qazi, A.K.; Mupparapu, N.; Guru, S.K.; Kumar, A.; Sharma, P.R.; Singh, S.K.; Singh, P.; Dar, M.J.; Bharate, S.B.; et al. Modulation of glycolysis and lipogenesis by novel PI3K selective molecule represses tumor angiogenesis and decreases colorectal cancer growth. Cancer Lett. 2016, 374, 250–260. [Google Scholar] [CrossRef]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprévote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc, L.; et al. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci. Rep. 2017, 7, 6945. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.E.; Short, S.P.; Williams, C.S. Colorectal Cancer and Metabolism. Curr. Colorectal Cancer Rep. 2018, 14, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, C.; Demuth, P.; Wittmann, S.; Fahrer, J. Targeting Altered Energy Metabolism in Colorectal Cancer: Oncogenic Reprogramming, the Central Role of the TCA Cycle and Therapeutic Opportunities. Cancers 2020, 12, 1731. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Fang, X. Advances in glucose metabolism research in colorectal cancer. Biomed. Rep. 2016, 5, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Zeindl-Eberhart, E.; Kirchner, T.; Jung, A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol. Res. Pract. 2009, 205, 858–862. [Google Scholar] [CrossRef]
- Ucar, G.; Ergun, Y.; Esen, S.A.; Acikgoz, Y.; Dirikoc, M.; Esen, İ.; Bal, Ö.; Uncu, D. Prognostic and predictive value of KRAS mutation number in metastatic colorectal cancer. Medicine 2020, 99, e22407. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Hou, M.; Guan, Y.-s.; Jiang, M.; Yang, Y.; Gou, H.-f. Expression of HIF-1alpha and VEGF in colorectal cancer: Association with clinical outcomes and prognostic implications. BMC Cancer 2009, 9, 432. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Li, J.; Sun, X.; Guo, Y.; Chu, D.; Wei, L.; Li, X.; Yang, G.; Liu, X.; Yao, L.; et al. Tumor suppressor NDRG2 inhibits glycolysis and glutaminolysis in colorectal cancer cells by repressing c-Myc expression. Oncotarget 2015, 6, 26161–26176. [Google Scholar] [CrossRef]
- Angius, A.; Uva, P.; Pira, G.; Muroni, M.R.; Sotgiu, G.; Saderi, L.; Uleri, E.; Caocci, M.; Ibba, G.; Cesaraccio, M.R.; et al. Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma. Int. J. Mol. Sci. 2019, 20, 4067. [Google Scholar] [CrossRef] [Green Version]
- Shiyong, L.; Bo, Y.; Ping, A.; Zhenjia, L.; Shujun, Y. Expression of thymidylate synthase gene and recurrence of colorectal carcinoma: Their relation and clinical significance. Chin. J. Surg. 2000, 38, 60–62. [Google Scholar]
- Houqiang, L.; Yinghao, Y. TS gene polymorphisms in gastric cancer and colorectal cancer. World Chin. J. Dig. 2013, 21, 865–872. [Google Scholar]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Khwairakpam, A.D.; Shyamananda, M.S.; Sailo, B.L.; Rathnakaram, S.R.; Padmavathi, G.; Kotoky, J.; Kunnumakkara, A.B. ATP citrate lyase (ACLY): A promising target for cancer prevention and treatment. Curr. Drug Targets 2015, 16, 156–163. [Google Scholar] [CrossRef]
- Zhan, Y.; Ginanni, N.; Tota, M.R.; Wu, M.; Bays, N.W.; Richon, V.M.; Kohl, N.E.; Bachman, E.S.; Strack, P.R.; Krauss, S. Control of cell growth and survival by enzymes of the fatty acid synthesis pathway in HCT-116 colon cancer cells. Clin. Cancer Res. 2008, 14, 5735–5742. [Google Scholar] [CrossRef] [Green Version]
- Pira, G.; Uva, P.; Scanu, A.M.; Rocca, P.C.; Murgia, L.; Uleri, E.; Piu, C.; Porcu, A.; Carru, C.; Manca, A.; et al. Landscape of transcriptome variations uncovering known and novel driver events in colorectal carcinoma. Sci. Rep. 2020, 10, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanankutty, A. PI3K/Akt/mTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. S-adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [Green Version]
- Zsigrai, S.; Kalmár, A.; Nagy, Z.B.; Barták, B.K.; Valcz, G.; Szigeti, K.A.; Galamb, O.; Dankó, T.; Sebestyén, A.; Barna, G.; et al. S-adenosylmethionine Treatment of Colorectal Cancer Cell Lines Alters DNA Methylation, DNA Repair and Tumor Progression-Related Gene Expression. Cells 2020, 9, 1864. [Google Scholar] [CrossRef]
- Bo, W.; He, J.; Ting, L.T.; Jie, L.; Qing, D.Y. Effects of methylation agent S-adenosyl-l-methionine on the biological behaviors of colorectal cancer cells. Guangdong Med. J. 2011, 32, 3157–3160. [Google Scholar]
- Mahmoud, A.M.; Ali, M.M. Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients 2019, 11, 608. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Prinz-Langenohl, R.; Fohr, I.; Pietrzik, K. Beneficial role for folate in the prevention of colorectal and breast cancer. Eur. J. Nutr. 2001, 40, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-I. Folate and DNA Methylation: A Mechanistic Link between Folate Deficiency and Colorectal Cancer? Cancer Epidemiol. Biomark. Prev. 2004, 13, 511–519. [Google Scholar]
- Duthie, S.J.; Narayanan, S.; Brand, G.M.; Pirie, L.; Grant, G. Impact of folate deficiency on DNA stability. J. Nutr. 2002, 132, 2444S–2449S. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J.; Narayanan, S.; Blum, S.; Pirie, L.; Brand, G.M. Folate deficiency in vitro induces uracil misincorporation and DNA hypomethylation and inhibits DNA excision repair in immortalized normal human colon epithelial cells. Nutr. Cancer 2000, 37, 245–251. [Google Scholar] [CrossRef]
- Song, J.; Sohn, K.J.; Medline, A.; Ash, C.; Gallinger, S.; Kim, Y.I. Chemopreventive effects of dietary folate on intestinal polyps in Apc+/− Msh2−/− mice. Cancer Res. 2000, 60, 3191–3199. [Google Scholar]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Chao, G.; Sheng-hao, W.; Wen-wei, Z.; Jin-hong, C.; Ming, L.; Lun-xiu, Q. Acetyl-CoA metabolism and cancer metastasis. Electron. J. Metab. Nutr. Cancer 2019, 6, 376–381. [Google Scholar]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Lin, S.-H.; Ren, F.; Li, J.-T.; Chen, J.-J.; Yao, C.-B.; Yang, H.-B.; Jiang, S.-X.; Yan, G.-Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Zhu, M.-J. Butyrate Inhibits Indices of Colorectal Carcinogenesis via Enhancing α-Ketoglutarate-Dependent DNA Demethylation of Mismatch Repair Genes. Mol. Nutr. Food Res. 2018, 62, e1700932. [Google Scholar] [CrossRef]
- Yun, Y.; Deyu, C. Research progress on acyl-CoA synthetase short chain family member 2 in tumorigenesis and development. J. New Med. 2021, 52, 234–238. [Google Scholar]
- Bae, J.M.; Kim, J.H.; Oh, H.J.; Park, H.E.; Lee, T.H.; Cho, N.-Y.; Kang, G.H. Downregulation of acetyl-CoA synthetase 2 is a metabolic hallmark of tumor progression and aggressiveness in colorectal carcinoma. Mod. Pathol. 2017, 30, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Luo, Y.; Li, L.; Tian, B.; Zheng, L.; Tian, T.; Yu, L.; Liao, X.; Zhan, W. Changes of histone acetylation modification levels in colorectal cancer. Chin. J. Gen. Surg. 2018, 27, 1348–1352. [Google Scholar]
- Guruswamy, S.; Swamy, M.V.; Choi, C.-I.; Steele, V.E.; Rao, C.V. S-adenosyl l-methionine inhibits azoxymethane-induced colonic aberrant crypt foci in F344 rats and suppresses human colon cancer Caco-2 cell growth in 3D culture. Int. J. Cancer 2008, 122, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, Y.-n.; Geng, X.; Zhang, W.-m. Experimental study on inhibition of colorectal cancer cell growth by S-adenosylmethionine. BME Clin. Med. 2011, 15, 183–186. [Google Scholar]
- Xiao, Y.; Su, X.; Huang, W.; Zhang, J.; Peng, C.; Huang, H.; Wu, X.; Huang, H.; Xia, M.; Ling, W. Role of S-adenosylhomocysteine in cardiovascular disease and its potential epigenetic mechanism. Int. J. Biochem. Cell Biol. 2015, 67, 158–166. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, L.; Zhong, T.; Mueller, M.; Men, Y.; Zhang, N.; Xie, J.; Giang, K.; Chung, H.; Sun, X.; et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat. Commun. 2015, 6, 10221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Feo, F. Chemoprevention of hepatocarcinogenesis: S-adenosyl-l-methionine. Alcohol 2002, 27, 193–198. [Google Scholar] [CrossRef]
- Garcea, R.; Daino, L.; Pascale, R.; Simile, M.M.; Puddu, M.; Ruggiu, M.E.; Seddaiu, M.A.; Satta, G.; Sequenza, M.J.; Feo, F. Protooncogene methylation and expression in regenerating liver and preneoplastic liver nodules induced in the rat by diethylnitrosamine: Effect of variations of S-adenosylmethionine: S-adenosylhomocysteine ratio. Carcinogenesis 1989, 10, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Sibani, S.; Melnyk, S.; Pogribny, I.P.; Wang, W.; Hiou-Tim, F.; Deng, L.; Trasler, J.; James, S.J.; Rozen, R. Studies of methionine cycle intermediates (SAM, SAH), DNA methylation and the impact of folate deficiency on tumor numbers in Min mice. Carcinogenesis 2002, 23, 61–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.I.; Salomon, R.N.; Graeme-Cook, F.; Choi, S.W.; Smith, D.E.; Dallal, G.E.; Mason, J.B. Dietary folate protects against the development of macroscopic colonic neoplasia in a dose responsive manner in rats. Gut 1996, 39, 732–740. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Herman, J.G.; Graff, J.R.; Vertino, P.M.; Issa, J.P. Alterations in DNA methylation: A fundamental aspect of neoplasia. Adv. Cancer Res. 1998, 72, 141–196. [Google Scholar]
- Schmutte, C.; Yang, A.S.; Nguyen, T.T.; Beart, R.W.; Jones, P.A. Mechanisms for the involvement of DNA methylation in colon carcinogenesis. Cancer Res. 1996, 56, 2375–2381. [Google Scholar]
- Lee, S.M.; Koh, H.-J.; Park, D.-C.; Song, B.J.; Huh, T.-L.; Park, J.-W. Cytosolic NADP(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic. Biol. Med. 2002, 32, 1185–1196. [Google Scholar] [CrossRef]
- Jia, P.; Kuang, X.; Wu, Y.; Sun, L.; Yuan, S. Progress on Relationship Between IDH Gene Mutation and Tumor. Prog. Mod. Biomed. 2019, 27, 1454–1457. [Google Scholar]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Gerecke, C.; Schumacher, F.; Berndzen, A.; Homann, T.; Kleuser, B. Vitamin C in combination with inhibition of mutant IDH1 synergistically activates TET enzymes and epigenetically modulates gene silencing in colon cancer cells. Epigenetics 2020, 15, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Ly, C.H.; Lynch, G.S.; Ryall, J.G. A Metabolic Roadmap for Somatic Stem Cell Fate. Cell Metab. 2020, 31, 1052–1067. [Google Scholar] [CrossRef]
- Wong, C.C.; Xu, J.; Li, W.; Qian, Y.; Kang, W.; Wu, J.; Sung, J.J.; Yu, J. Slc25A22 Drives Oncometabolite Succinate to Promote Wnt/Β-Catenin Signaling and Cancer Stemness in Kras-Mutant Colorectal Cancer. Gastroenterology 2019, 156, S-188. [Google Scholar] [CrossRef]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangaraju, M.; Carswell, K.N.; Prasad, P.D.; Ganapathy, V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem. J. 2009, 417, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cao, L.; Tian, Y.; Zhang, P.; Ding, C.; Lu, W.; Jia, C.; Shao, C.; Liu, W.; Wang, D.; et al. Butyrate Suppresses the Proliferation of Colorectal Cancer Cells via Targeting Pyruvate Kinase M2 and Metabolic Reprogramming. Mol. Cell. Proteom. 2018, 17, 1531–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.M.; Hwang, S.W.; Wang, T.; Park, C.W.; Ryu, Y.-M.; Jung, J.-H.; Shin, J.H.; Kim, S.-Y.; Lee, J.L.; Kim, C.W.; et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species level. Cancer Sci. 2019, 110, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Brandl, L.; Kirstein, N.; Neumann, J.; Sendelhofert, A.; Vieth, M.; Kirchner, T.; Menssen, A. The c-MYC/NAMPT/SIRT1 feedback loop is activated in early classical and serrated route colorectal cancer and represents a therapeutic target. Med. Oncol. 2018, 36, 5. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Hua, L. Expression of SIRT6 Protein in Colon Cancer and Its Effect on the Biological Behavior of Colon Cancer Cells. Master’s Thesis, Jinzhou Medical University, Jinzhou, China, 2019. [Google Scholar]
- Wang, J.; Zhang, M.; Zhang, D.; Fan, Q. Effects of SIRT5 Gene Silencing on Migration and Invasion of Colonic Carcinoma Cells. Mil. Med. J. S Chin. 2017, 31, 225–228. [Google Scholar]
- Qin, Y.; Jiang, M.; Feng, J.; Bian, X.; Xu, H. The Emerging Roles of SIRT7 in Tumorigenesis. Prog. Mod. Biomed. 2017, 17, 1964–1967. [Google Scholar]
- Wang, B.; Shen, Z.; Ye, Y.; Jiang, K.; Wang, S. SIRT2 expression in colorectal cancer patients with liver metastasis and its clinical significance. Chin. J. Gen. Surg. 2019, 12, 49–52. [Google Scholar]
- Huang, G. The Expression of SIRT4 in Gastrointestinal Carcinoma and Its Effect on Human Colorectal Cancer Cell Strains’ Biological Behavior Study. Ph.D. Thesis, Shanghai Jiaotong University, Shanghai, China, 2015. [Google Scholar]
- Wei, Z.; Song, J.; Wang, G.; Cui, X.; Zheng, J.; Tang, Y.; Chen, X.; Li, J.; Cui, L.; Liu, C.-Y.; et al. Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat. Commun. 2018, 9, 4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Wei, P.; Wu, J.; Zhang, M.; Li, G.; Li, Y.; Xu, Y.; Li, X.; Xie, D.; Cai, S.; et al. The FOXC1/FBP1 signaling axis promotes colorectal cancer proliferation by enhancing the Warburg effect. Oncogene 2019, 38, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Li, N.; Qian, Z.; Zhu, M.; Li, Q.; Zheng, J.; Wang, X.; Shi, G. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS ONE 2011, 6, e25564. [Google Scholar] [CrossRef]
- Desai, S.; Ding, M.; Wang, B.; Lu, Z.; Zhao, Q.; Shaw, K.; Yung, W.K.A.; Weinstein, J.N.; Tan, M.; Yao, J. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget 2014, 5, 8202–8210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, P.-H.; Hwang, J.-H.; Kwak, D.-K.; Koh, E.; Kim, K.-S.; Choi, K.-Y. APC loss induces Warburg effect via increased PKM2 transcription in colorectal cancer. Br. J. Cancer 2021, 124, 634–644. [Google Scholar] [CrossRef]
- Katagiri, M.; Karasawa, H.; Takagi, K.; Nakayama, S.; Yabuuchi, S.; Fujishima, F.; Naitoh, T.; Watanabe, M.; Suzuki, T.; Unno, M.; et al. Hexokinase 2 in colorectal cancer: A potent prognostic factor associated with glycolysis, proliferation and migration. Histol. Histopathol. 2017, 32, 351–360. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Zhang, Y.H.; Qin, X.J.; Wang, Y.X.; Fu, J. Circular RNA circDENND4C facilitates proliferation, migration and glycolysis of colorectal cancer cells through miR-760/GLUT1 axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2387–2400. [Google Scholar] [CrossRef]
- Du, L.; Duan, W.; Jiang, X.; Zhao, L.; Li, J.; Wang, R.; Yan, S.; Xie, Y.; Yan, K.; Wang, Q.; et al. Cell-free lncRNA expression signatures in urine serve as novel non-invasive biomarkers for diagnosis and recurrence prediction of bladder cancer. J. Cell. Mol. Med. 2018, 22, 2838–2845. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, Z.; Zou, K.; Cheng, Y.; Yang, M.; Chen, H.; Wang, H.; Zhao, J.; Chen, P.; He, L.; et al. MiR-1 suppresses tumor cell proliferation in colorectal cancer by inhibition of Smad3-mediated tumor glycolysis. Cell Death Dis. 2017, 8, e2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregersen, L.H.; Jacobsen, A.; Frankel, L.B.; Wen, J.; Krogh, A.; Lund, A.H. MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer 2012, 12, 232. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Sakai, M.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakayama, T.; Ueda, H.; Nakagawa, Y.; Ito, Y.; et al. PTBP1-associated microRNA-1 and -133b suppress the Warburg effect in colorectal tumors. Oncotarget 2016, 7, 18940–18952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Hou, L.; Li, L.; Li, L.; Zhu, L.; Wang, Y.; Huang, X.; Hou, Y.; Zhu, D.; Zou, H.; et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene 2020, 39, 469–485. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, N. MiR-335-5p Inhibits Cell Proliferation, Migration and Invasion in Colorectal Cancer through Downregulating LDHB. J. BUON 2019, 24, 1128–1136. [Google Scholar] [PubMed]
- Wang, J.; Wang, H.; Liu, A.; Fang, C.; Hao, J.; Wang, Z. Lactate dehydrogenase A negatively regulated by miRNAs promotes aerobic glycolysis and is increased in colorectal cancer. Oncotarget 2015, 6, 19456–19468. [Google Scholar] [CrossRef]
- Lan, Z.; Yao, X.; Sun, K.; Li, A.; Liu, S.; Wang, X. The Interaction Between lncRNA SNHG6 and hnRNPA1 Contributes to the Growth of Colorectal Cancer by Enhancing Aerobic Glycolysis Through the Regulation of Alternative Splicing of PKM. Front. Oncol. 2020, 10, 363. [Google Scholar] [CrossRef]
- Bian, Z.; Zhang, J.; Li, M.; Feng, Y.; Wang, X.; Zhang, J.; Yao, S.; Jin, G.; Du, J.; Han, W.; et al. LncRNA-FEZF1-AS1 Promotes Tumor Proliferation and Metastasis in Colorectal Cancer by Regulating PKM2 Signaling. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4808–4819. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wei, M.; Wang, C.; Sun, D.; Liu, P.; Zhong, X.; Yu, W. Long noncoding RNA KCNQ1OT1 promotes colorectal carcinogenesis by enhancing aerobic glycolysis via hexokinase-2. Aging (Albany NY) 2020, 12, 11685–11697. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, P.; Du, J.; Chen, J.; Liu, W.; Ye, K. LncRNA RAD51-AS1/miR-29b/c-3p/NDRG2 crosstalk repressed proliferation, invasion and glycolysis of colorectal cancer. IUBMB Life 2021, 73, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Shen, C.; Jiang, P.; Yu, C.; Guo, F.; Tian, X.; Zhu, X.; Lu, S.; Han, B.; Zhong, M.; et al. Risk SNP-induced lncRNA-SLCC1 drives colorectal cancer through activating glycolysis signaling. Signal Transduct. Target. 2021, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004, 64, 3014–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trojan, J.; Brieger, A.; Raedle, J.; Esteller, M.; Zeuzem, S. 5′-CpG island methylation of the LKB1/STK11 promoter and allelic loss at chromosome 19p13.3 in sporadic colorectal cancer. Gut 2000, 47, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Rawluszko, A.A.; Bujnicka, K.E.; Horbacka, K.; Krokowicz, P.; Jagodziński, P.P. Expression and DNA methylation levels of prolyl hydroxylases PHD1, PHD2, PHD3 and asparaginyl hydroxylase FIH in colorectal cancer. BMC Cancer 2013, 13, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Chen, L.; Yang, J.; Ye, T.; Chen, Y.; Fang, J. HIF-1α-induced histone demethylase JMJD2B contributes to the malignant phenotype of colorectal cancer cells via an epigenetic mechanism. Carcinogenesis 2012, 33, 1664–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Cui, G.; Tang, C.-W.; Zhang, X.-L.; Dai, C.; Xu, Y.-Q.; Gong, H.; Xue, T.; Guo, H.-H.; Bao, Y. Role of glucose metabolism related gene GLUT1 in the occurrence and prognosis of colorectal cancer. Oncotarget 2017, 8, 56850–56857. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Serra, P.; Marcilla, M.; Villanueva, A.; Ramos-Fernandez, A.; Palau, A.; Leal, L.; Wahi, J.E.; Setien-Baranda, F.; Szczesna, K.; Moutinho, C.; et al. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat. Commun. 2014, 5, 3608. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Chen, H.; Ye, J.; Wei, X.; Liu, S.; Wang, R. HIF-1α/Ascl2/miR-200b regulatory feedback circuit modulated the epithelial-mesenchymal transition (EMT) in colorectal cancer cells. Exp. Cell Res. 2017, 360, 243–256. [Google Scholar] [CrossRef]
- Chen, T.; Yao, L.-Q.; Shi, Q.; Ren, Z.; Ye, L.-C.; Xu, J.-M.; Zhou, P.-H.; Zhong, Y.-S. MicroRNA-31 contributes to colorectal cancer development by targeting factor inhibiting HIF-1α (FIH-1). Cancer Biol. 2014, 15, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhao, J.; Xu, J.; Wang, J.; Jia, J. miR-526b-3p functions as a tumor suppressor in colon cancer by regulating HIF-1α. Am. J. Transl. Res. 2016, 8, 2783–2789. [Google Scholar]
- Yang, L.G.; Cao, M.Z.; Zhang, J.; Li, X.Y.; Sun, Q.L. LncRNA XIST modulates HIF-1A/AXL signaling pathway by inhibiting miR-93-5p in colorectal cancer. Mol. Genet. Genom. Med. 2020, 8, e1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shi, H.; Tang, H.; Fang, Z.; Wang, J.; Cui, S. miR-218 inhibits the invasion and migration of colon cancer cells by targeting the PI3K/Akt/mTOR signaling pathway. Int. J. Mol. Med. 2015, 35, 1301–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, G.; Yao, X.; Zhang, Y.; Gu, J.; Geng, Y.; Xue, F.; Zhang, J. Colorectal cancer cell-derived exosomes containing miR-10b regulate fibroblast cells via the PI3K/Akt pathway. Bull. Cancer 2018, 105, 336–349. [Google Scholar] [CrossRef]
- Tang, J.; Yan, T.; Bao, Y.; Shen, C.; Yu, C.; Zhu, X.; Tian, X.; Guo, F.; Liang, Q.; Liu, Q.; et al. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-Myc. Nat. Commun. 2019, 10, 3499. [Google Scholar] [CrossRef]
- Feng, J.; Ma, J.; Liu, S.; Wang, J.; Chen, Y. A noncoding RNA LINC00504 interacts with c-Myc to regulate tumor metabolism in colon cancer. J. Cell Biochem. 2019, 120, 14725–14734. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, J.H.; Wu, Q.N.; Jin, Y.; Wang, D.S.; Chen, Y.X.; Liu, J.; Luo, X.J.; Meng, Q.; Pu, H.Y.; et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol. Cancer 2019, 18, 174. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yang, W.; Li, N.; Chen, X.; Ma, F.; Yang, J.; Zhang, Y.; Chai, X.; Zhang, B.; Hou, X.; et al. LncRNA MCF2L-AS1 aggravates proliferation, invasion and glycolysis of colorectal cancer cells via the crosstalk with miR-874-3p/FOXM1 signaling axis. Carcinogenesis 2021, 42, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhu, K.; Liu, L.; Gu, J.; Niu, H.; Guo, J. lncARSR sponges miR-34a-5p to promote colorectal cancer invasion and metastasis via hexokinase-1-mediated glycolysis. Cancer Sci. 2020, 111, 3938–3952. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Yang, H.; Bai, M.; Ning, T.; Deng, T.; Liu, R.; Fan, Q.; Zhu, K.; Li, J.; et al. Exosome-delivered circRNA promotes glycolysis to induce chemoresistance through the miR-122-PKM2 axis in colorectal cancer. Mol. Oncol. 2020, 14, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, Y.; Liu, L.; Yang, W.; Zhang, Q. HNF1A-AS1 Regulates Cell Migration, Invasion and Glycolysis via Modulating miR-124/MYO6 in Colorectal Cancer Cells. OncoTargets Ther. 2020, 13, 1507–1518. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, H.; Ye, X.; Ji, A.; Fu, X.; Wu, H.; Zeng, X. Hsa_circ_0000231 knockdown inhibits the glycolysis and progression of colorectal cancer cells by regulating miR-502-5p/MYO6 axis. World J. Surg. Oncol. 2020, 18, 255. [Google Scholar] [CrossRef]
- Li, Z.; Yao, H.; Wang, S.; Li, G.; Gu, X. CircTADA2A suppresses the progression of colorectal cancer via miR-374a-3p/KLF14 axis. J. Exp. Clin. Cancer Res. 2020, 39, 160. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-S.; Little, J.B.; Yuan, Z.-M. Glycolytic metabolism influences global chromatin structure. Oncotarget 2015, 6, 4214–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liu, Z.; Zou, X.; Lan, Y.; Sun, X.; Wang, X.; Zhao, S.; Jiang, C.; Liu, H. Mechanisms underlying 3-bromopyruvate-induced cell death in colon cancer. J. Bioenerg. Biomembr. 2015, 47, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Song, P.; Zhu, L.; Aziz, N.; Zhou, Q.; Zhang, Y.; Xu, W.; Feng, L.; Chen, D.; Wang, X.; et al. Synthetic lethality of glutaminolysis inhibition, autophagy inactivation and asparagine depletion in colon cancer. Oncotarget 2017, 8, 42664–42672. [Google Scholar] [CrossRef] [Green Version]
- Glazer, R.I.; Knode, M.C.; Tseng, C.K.; Haines, D.R.; Marquez, V.E. 3-Deazaneplanocin A: A new inhibitor of S-adenosylhomocysteine synthesis and its effects in human colon carcinoma cells. Biochem. Pharm. 1986, 35, 4523–4527. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Han, Z.; Zhao, N.; Zhu, B.; Zhang, Q.; Yang, X.; Sheng, D.; Hou, J.; Guo, S.; Wei, L.; et al. Inhibition of DNMT suppresses the stemness of colorectal cancer cells through down-regulating Wnt signaling pathway. Cell. Signal. 2018, 47, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Yoo, C.B.; Weisenberger, D.J.; Chuang, J.; Wozniak, C.; Liang, G.; Marquez, V.E.; Greer, S.; Orntoft, T.F.; Thykjaer, T.; et al. Preferential response of cancer cells to zebularine. Cancer Cell 2004, 6, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Chen, B.; Qin, S.; Li, S.; He, X.; Qiu, S.; Zhao, W.; Zhao, H. A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem. Biophys. Res. Commun. 2010, 392, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.C.; Petersen, J.; Nielsen, S.J.; Morsczeck, C.; Morszeck, C.; Jensen, P.B.; Sehested, M.; Grauslund, M. Proteomic profiling of human colon cancer cells treated with the histone deacetylase inhibitor belinostat. Electrophoresis 2010, 31, 2714–2721. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.-P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Endo, S.; Saito, R.; Hirose, M.; Hirai, S.; Suzuki, H.; Yamato, K.; Hyodo, I. The sirtuin inhibitor tenovin-6 upregulates death receptor 5 and enhances cytotoxic effects of 5-fluorouracil and oxaliplatin in colon cancer cells. Oncol. Res. 2013, 21, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Feldman, J.L.; Baeza, J.; Denu, J.M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Xu, J.; Li, Y.; Zhao, R.; Du, S.; Lv, C.; Wu, W.; Liu, R.; Sheng, X.; Song, Y.; et al. MicroRNA-31 Reduces Inflammatory Signaling and Promotes Regeneration in Colon Epithelium, and Delivery of Mimics in Microspheres Reduces Colitis in Mice. Gastroenterology 2019, 156, 2281–2296. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, L.N.; Xie, Y. MiRNA-802 inhibits the metastasis of colorectal cancer by targeting FOXE1. Eur. Rev. Med. Pharm. Sci. 2020, 24, 1778–1785. [Google Scholar] [CrossRef]
- Valeri, N.; Braconi, C.; Gasparini, P.; Murgia, C.; Lampis, A.; Paulus-Hock, V.; Hart, J.R.; Ueno, L.; Grivennikov, S.I.; Lovat, F.; et al. MicroRNA-135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell 2014, 25, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Jüttermann, R.; Li, E.; Jaenisch, R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA 1994, 91, 11797–11801. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, A.S.; Hong, J.Y.; Cao, J.; Lu, X.; Price, I.R.; Zhao, Q.; Kosciuk, T.; Yang, M.; Bai, J.J.; Lin, H. Novel Lysine-Based Thioureas as Mechanism-Based Inhibitors of Sirtuin 2 (SIRT2) with Anticancer Activity in a Colorectal Cancer Murine Model. J. Med. Chem. 2019, 62, 4131–4141. [Google Scholar] [CrossRef]
- Dong, Z.; Cui, H. Function of Sirtuins in Cancer Stem Cells. Int. J. Stem Cell Res. Ther. 2016, 3, 24. [Google Scholar] [CrossRef]
- van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseinahli, N.; Aghapour, M.; Duijf, P.H.G.; Baradaran, B. Treating cancer with microRNA replacement therapy: A literature review. J. Cell. Physiol. 2018, 233, 5574–5588. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.-S.; Jung, K.-A.; Kim, S.-M.; Hong, Y.S.; Ryu, M.-H.; Jang, S.J.; Moon, D.H.; Cho, D.-H.; Kim, J.C.; Lee, J.S.; et al. The histone deacetylase inhibitor PXD101 increases the efficacy of irinotecan in in vitro and in vivo colon cancer models. Cancer Chemother. Pharm. 2011, 68, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L.; Cleris, L.; Magistroni, V.; Piazza, R.; Boschelli, F.; Formelli, F.; Gambacorti-Passerini, C. Valproic acid enhances bosutinib cytotoxicity in colon cancer cells. Int. J. Cancer 2009, 124, 1990–1996. [Google Scholar] [CrossRef]
- Raskov, H.; Søby, J.H.; Troelsen, J.; Bojesen, R.D.; Gögenur, I. Driver Gene Mutations and Epigenetics in Colorectal Cancer. Ann. Surg. 2020, 271, 75–85. [Google Scholar] [CrossRef]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 3763. [Google Scholar] [CrossRef]
- Devarasetty, M.; Skardal, A.; Cowdrick, K.; Marini, F.; Soker, S. Bioengineered Submucosal Organoids for In Vitro Modeling of Colorectal Cancer. Tissue Eng. Part A 2017, 23, 1026–1041. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Inhibitor | Target Enzyme | Mode of Action | Ongoing Clinical Use/Trials | Ref. |
|---|---|---|---|---|
| 2-Deoxyglucose (2-DG) | Hexokinases | It has a structure similar to glucose and can competitively bind to HK2 with glucose; it can also inhibit acetyl-CoA and increase the activity of HDACs | Phase I/II for prostate cancer, Phase I dose escalation trial | [188] |
| 3-Bromopyruvate (3-BrPA) | Hexokinases | It can inhibit the activity of HK2, thereby inhibiting the production of ATP and inducing the death of CRC cells | NA | [189] |
| Compound 968 | Glutaminases | It can inhibit the recombinant expression of GLS1 and combine with inactivated GLS to prevent GLS1 activation | NA | [190] |
| 3-deazaneplanocin A (DZNep) | SAH hydrolase | It suppresses DNA and histone methylation by reducing the SAM/SAH ratio in CRC | NA | [191] |
| Inhibitor | Target Enzyme | Mode of Action | Ongoing Clinical Use/Trials | Ref. |
|---|---|---|---|---|
| DNMT inhibitors | ||||
| 5-Azacytidine, 5-Aza-2′-deoxycytidine, Zebularine | DNA methyltransferases | The first two drugs inactivate DNMTs non-selectively. Unlike the first two, zebularine has much smaller side effects and shows high selectivity in inhibiting DNMTs | 5-Azacytidine (Phase I–III for various malignant tumors; Phase II for metastatic CRC), 5-Aza-2′-deoxycytidine (Phase I–III for various malignant tumors; Phase I for liver metastatic CRC;) | [192,193] |
| HDAC inhibitors | ||||
| Chidamide, Belinostat, Valproic acid | Histone deacetylases | HDAC inhibitors can induce histone acetylation and reverse the abnormal gene expression caused by HDACs | Chidamide, Valproic acid (Phase I–III for various malignant tumors; Phase II for metastatic CRC), Belinostat (Phase I/II for various malignant tumors; Phase I for CRC) | [194,195,196] |
| SIRT activators and inhibitors | ||||
| Tenovin-6 | SIRT1, SIRT2 | It inhibits the protein deacetylation activity of SIRT1 and SIRT2 | NA | [197] |
| Myristic acid, Oleic acid, Linoleic acid | SIRT6 | Free fatty acids activate SIRT6, which functions as a tumor suppressor to inhibit glycolysis | NA | [198] |
| miRNA modulators | ||||
| miRNA mimics, miRNAs in encoding expression vectors | miRNAs | They can effectively restore the functions of these “lost” miRNAs. For example, restoring the down-regulated expression of miR-31 can inhibit the proliferation, migration and invasion of CRC cells. | miR-16 mimic (Phase I for non-small cell lung cancer) | [160,199,200] |
| Antisense nucleotides, miRNA sponges | miRNAs | They can inhibit miRNAs that are overexpressed in tumors. For example, specific inhibition of miR-135b can inhibit the proliferation, migration and induce apoptosis of CRC cells. | NA | [201] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Dong, Z.; Cui, H. Interplay between Epigenetics and Cellular Metabolism in Colorectal Cancer. Biomolecules 2021, 11, 1406. https://doi.org/10.3390/biom11101406
Zhang X, Dong Z, Cui H. Interplay between Epigenetics and Cellular Metabolism in Colorectal Cancer. Biomolecules. 2021; 11(10):1406. https://doi.org/10.3390/biom11101406
Chicago/Turabian StyleZhang, Xiaolin, Zhen Dong, and Hongjuan Cui. 2021. "Interplay between Epigenetics and Cellular Metabolism in Colorectal Cancer" Biomolecules 11, no. 10: 1406. https://doi.org/10.3390/biom11101406
APA StyleZhang, X., Dong, Z., & Cui, H. (2021). Interplay between Epigenetics and Cellular Metabolism in Colorectal Cancer. Biomolecules, 11(10), 1406. https://doi.org/10.3390/biom11101406