
An Insight of RuBisCO Evolution through a Multilevel Approach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Classification of RuBisCO Isoforms
2.2. RuBisCO Structure Selection and Phylogenetic Analysis
2.3. Normal Mode Analysis
2.4. Molecular Dynamics
2.5. Stability and Flexibility Analysis
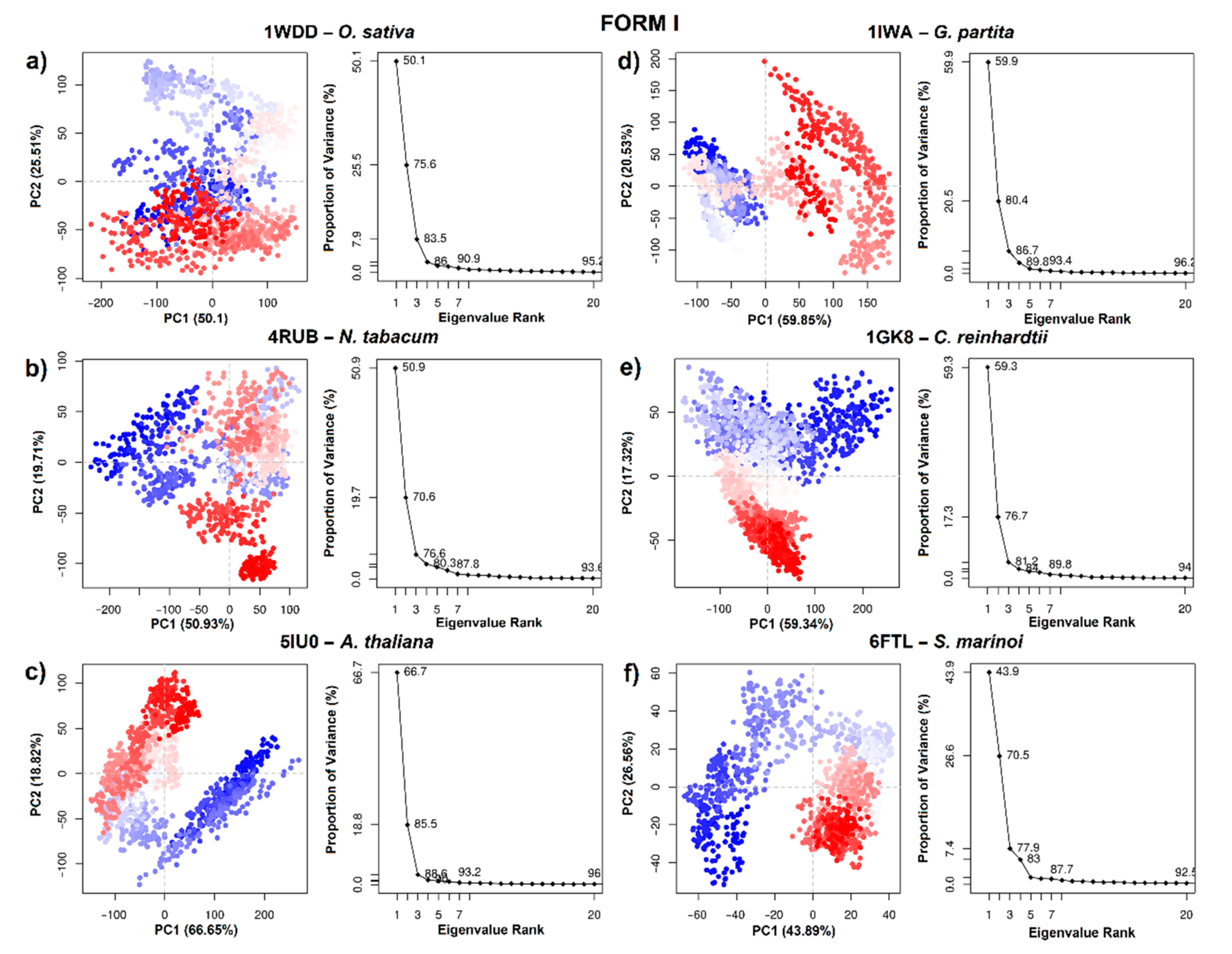
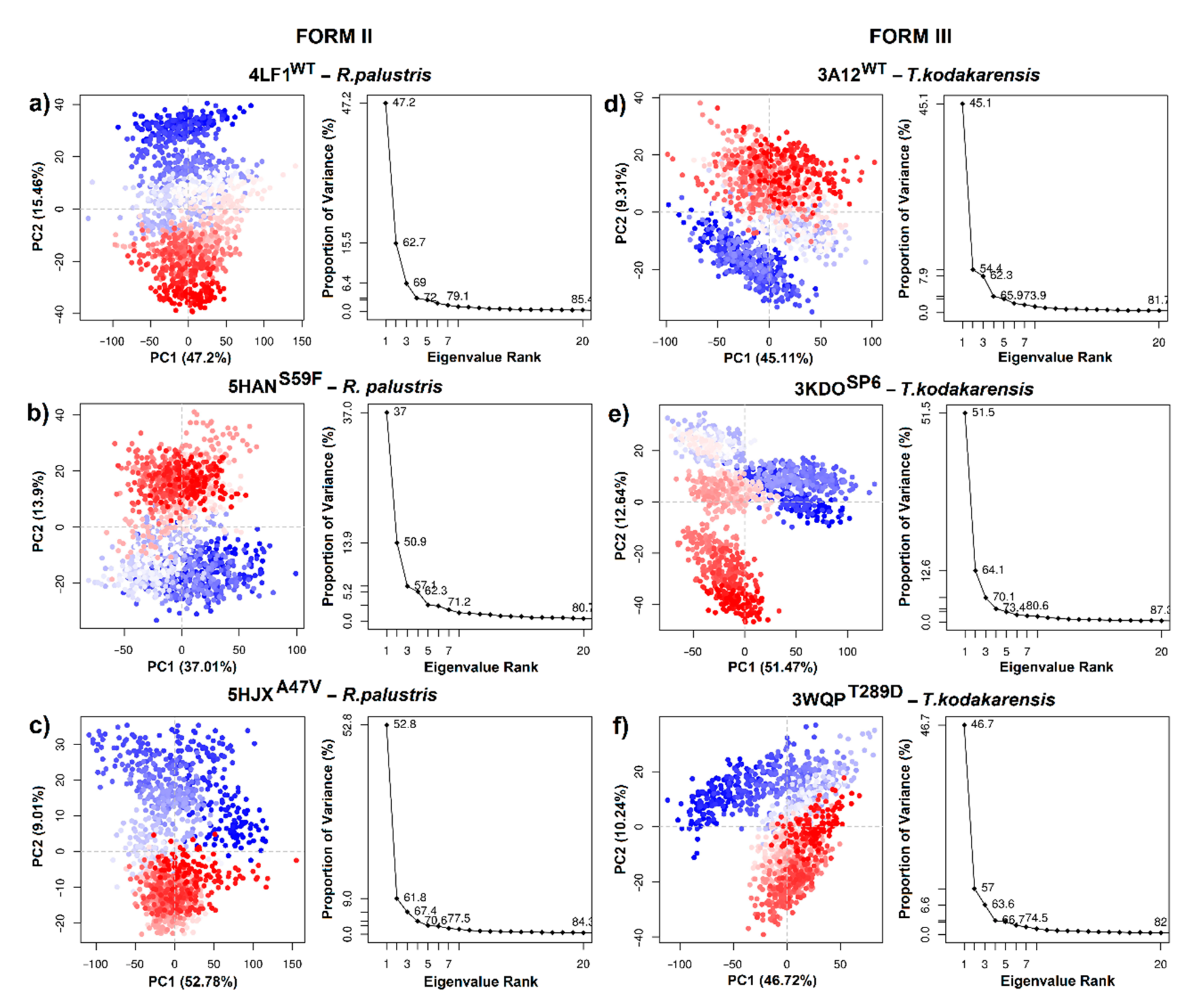
2.6. Principal Component Analysis
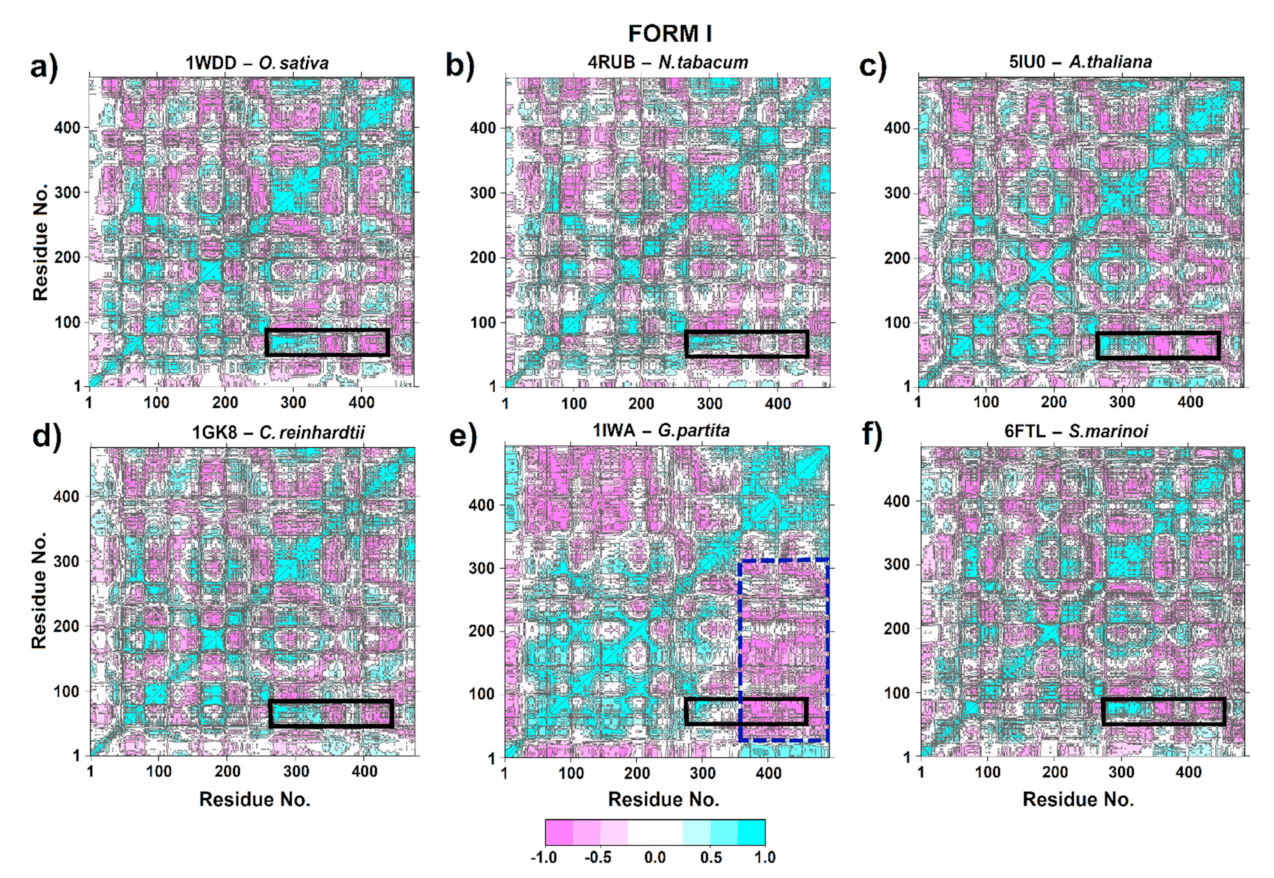
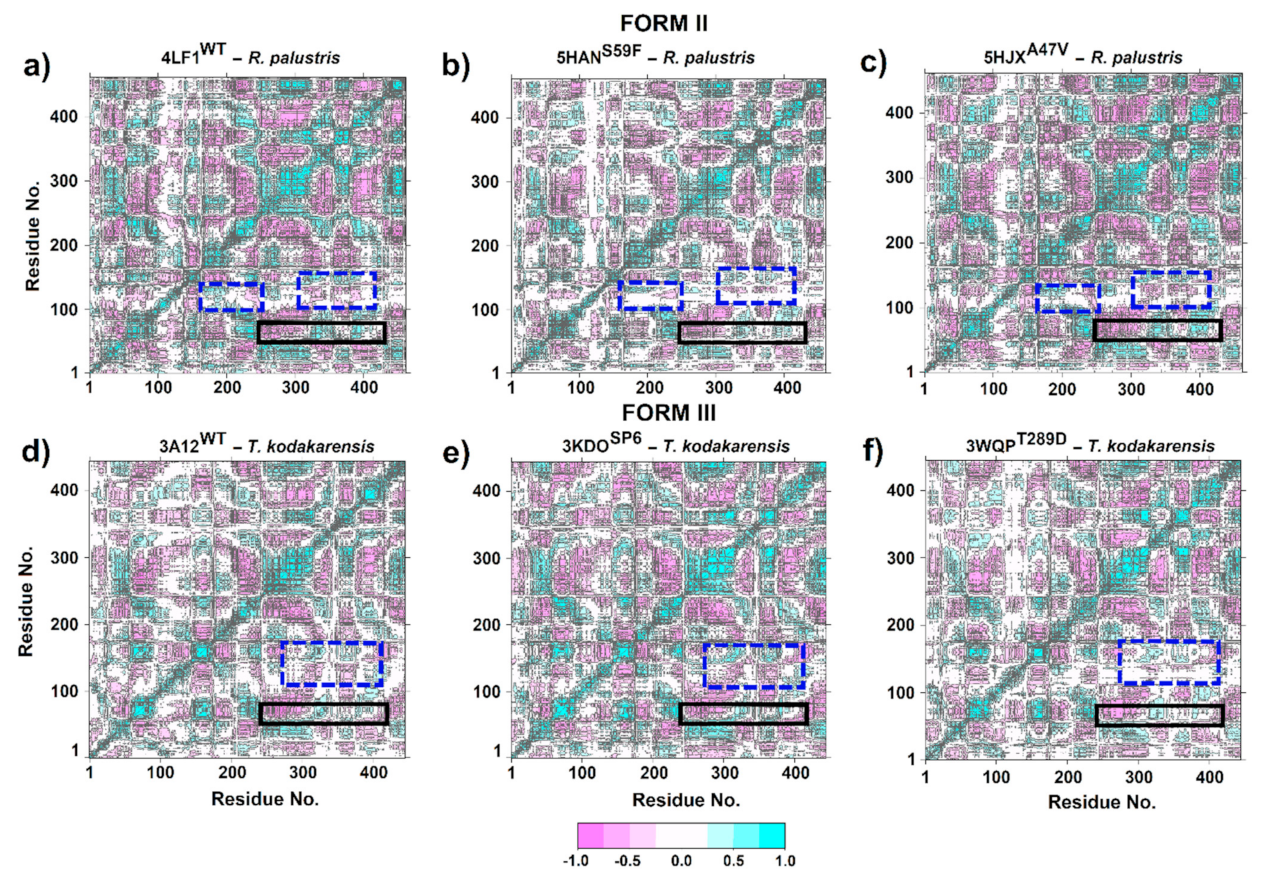
2.7. Dynamic Cross-Correlation Matrices (DCCM)
3. Results
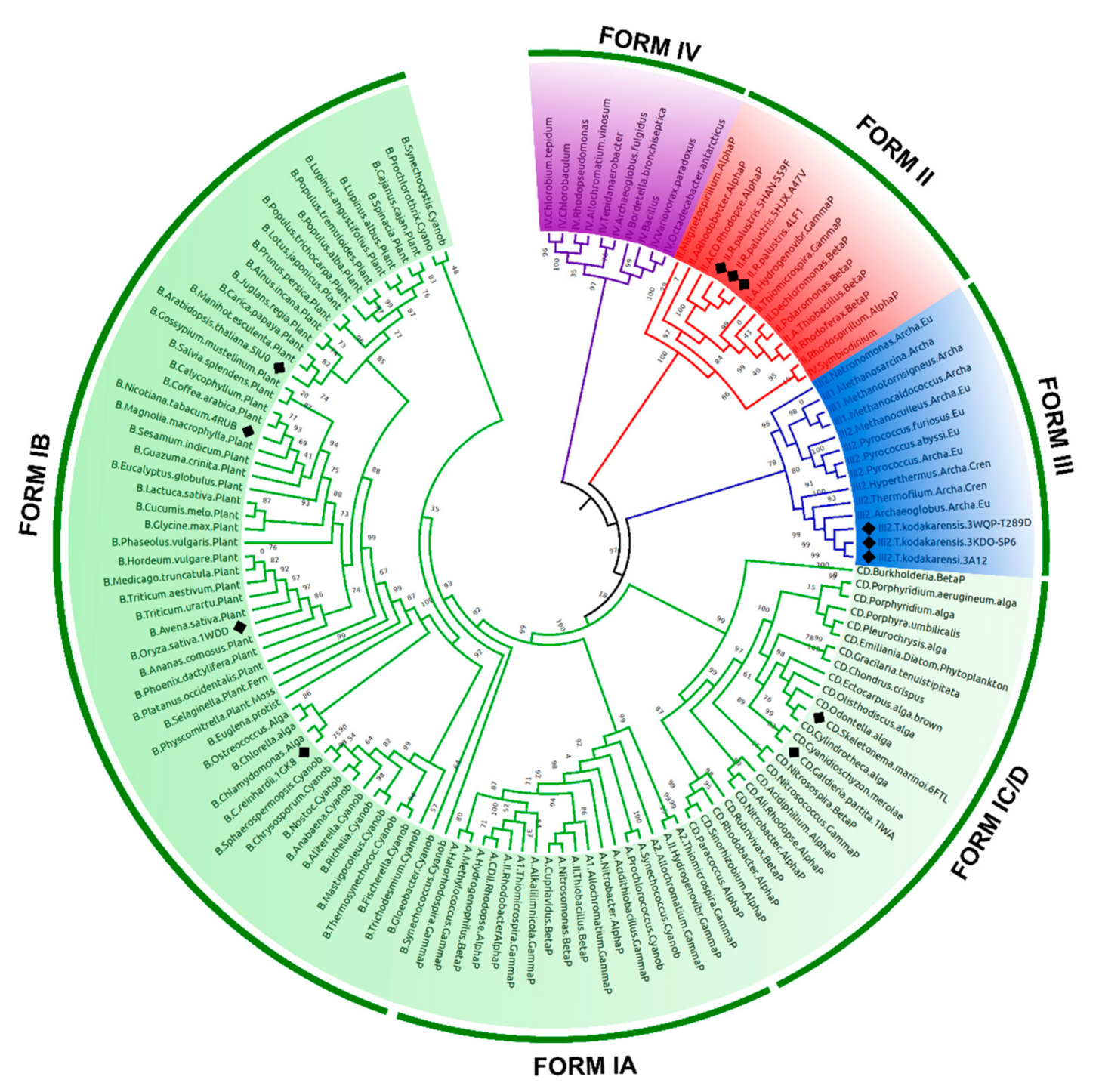
3.1. RuBisCO Forms Classification
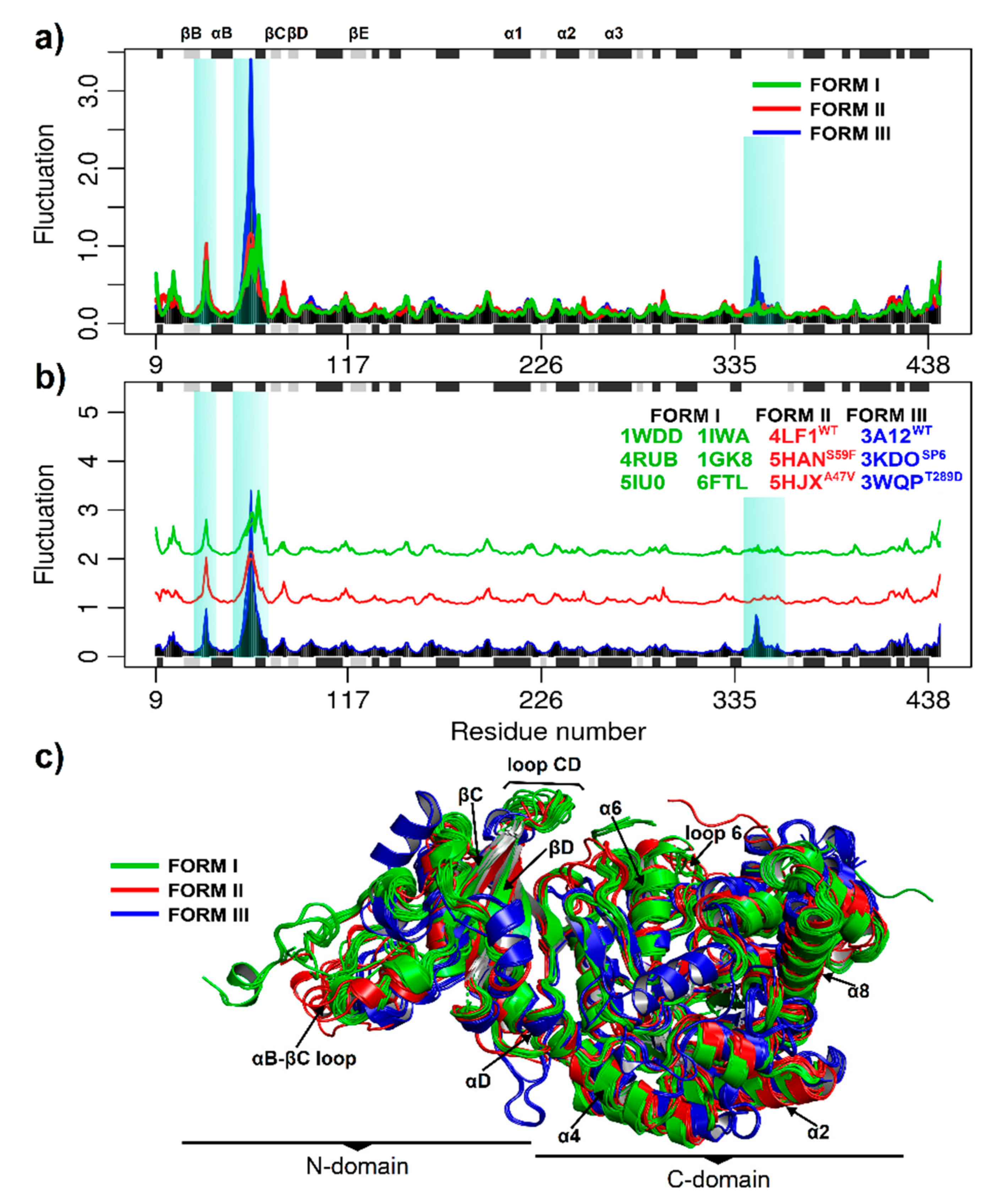
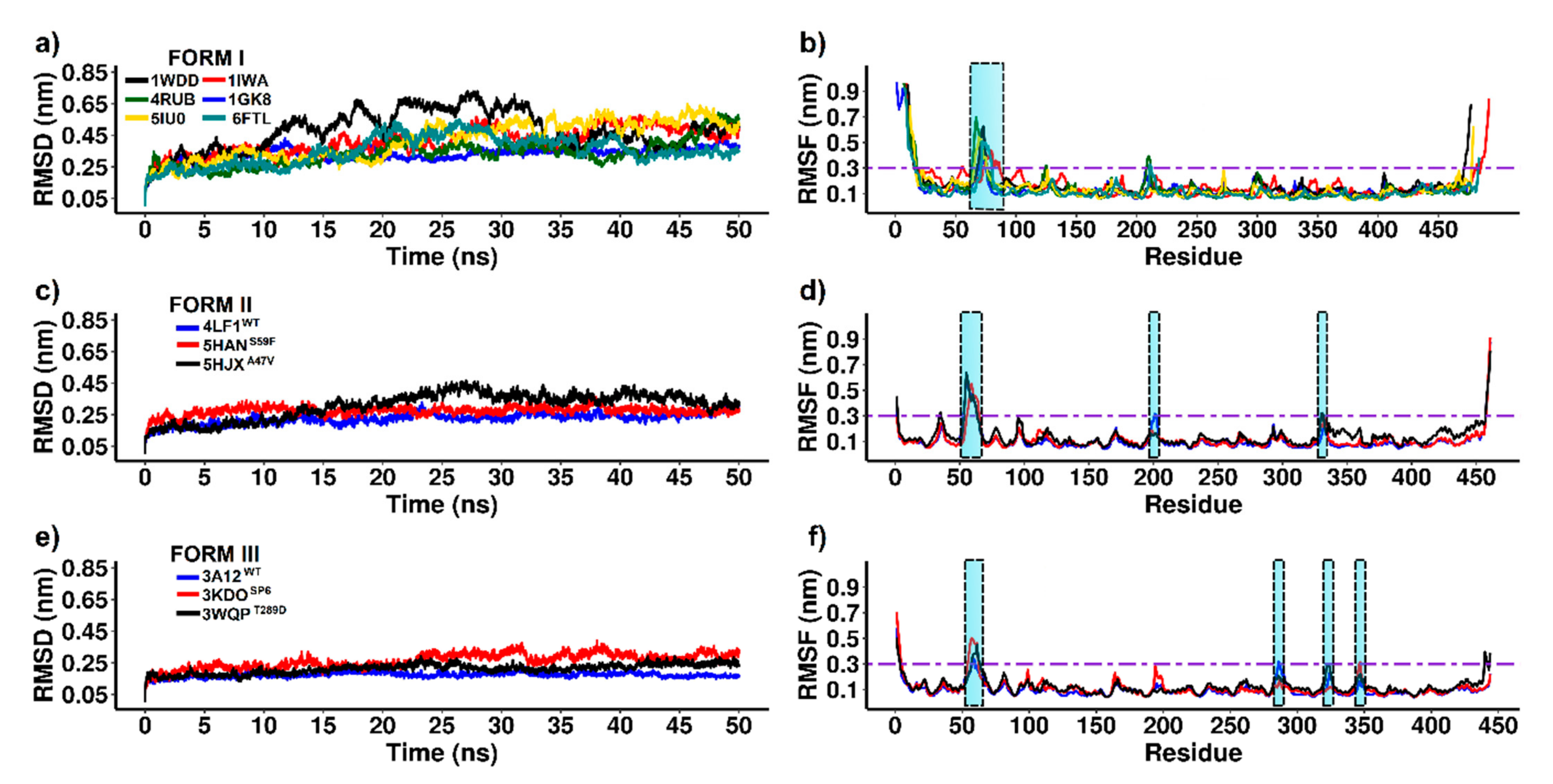
3.2. Stability and Flexibility Evaluation of RuBisCO Forms
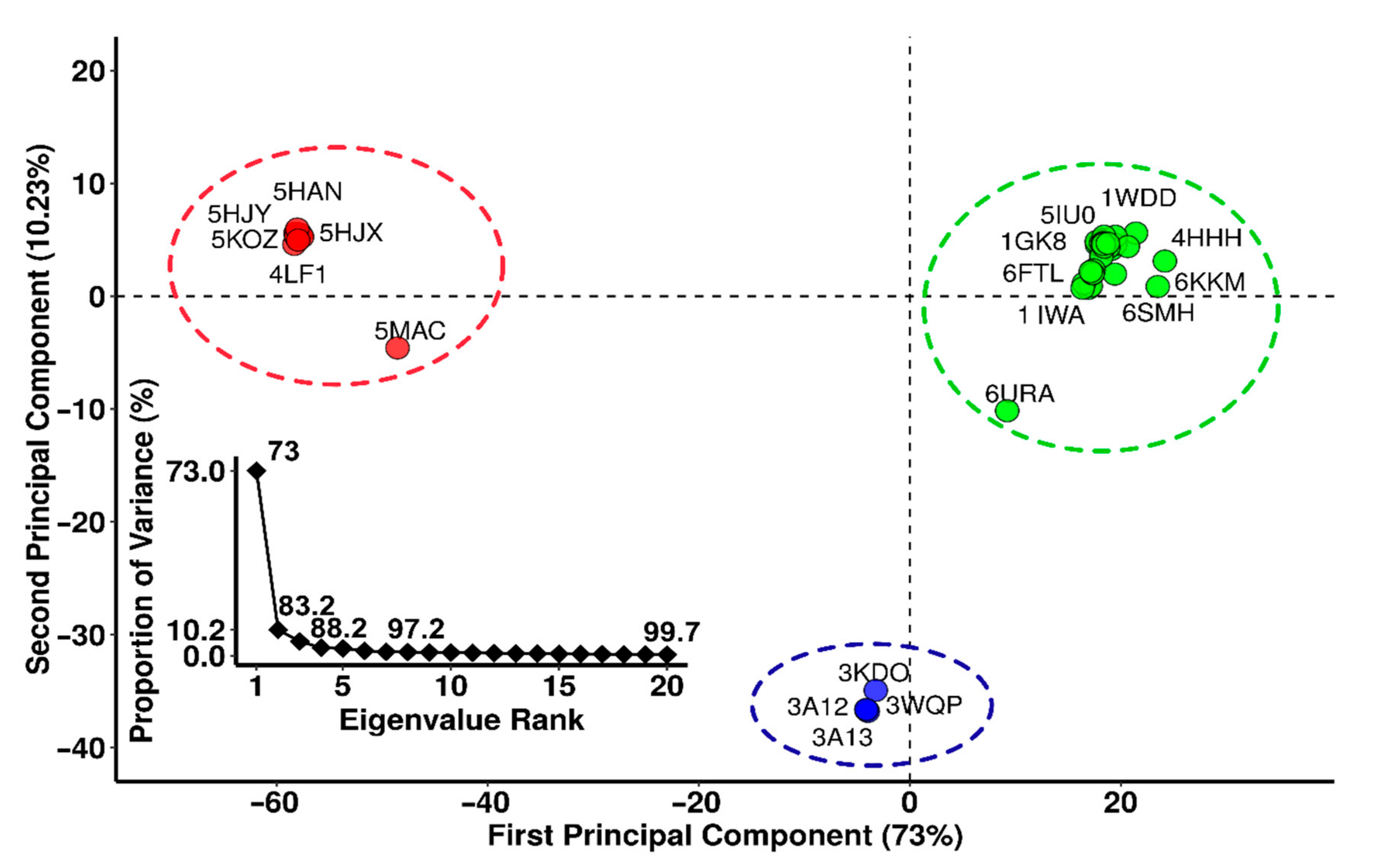
3.3. Principal Component Analysis (PCA)
3.4. Dynamic Cross-Correlation Matrix (DCCM)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersson, I.; Backlund, A. Structure and Function of Rubisco. Plant Phys. Biochem. 2008, 46, 275–291. [Google Scholar] [CrossRef]
- Tabita, F.R.; Hanson, T.E.; Li, H.; Satagopan, S.; Singh, J.; Chan, S. Function, Structure, and Evolution of the RubisCO-Like Proteins and Their RubisCO Homologs. Microbiol. Mol. Biol. Rev. 2007, 71, 576–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erb, T.; Zarzycki, J. A Short History of RubisCO: The Rise and Fall (?) of Nature’s Predominant CO2 Fixing Enzyme. Plant Biotechnol. 2018, 49, 100–107. [Google Scholar] [CrossRef]
- Stec, B. Structural Mechanism of RuBisCO Activation by Carbamylation of the Active Site Lysine. Proc. Natl. Acad. Sci. USA 2012, 109, 18785–18790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Chettiyan, R.; Ramya, S.; Mueller-cajar, O. Surveying the Expanding Prokaryotic Rubisco Multiverse. FEMS Microbiol. Lett. 2017, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitano, K.; Maeda, N.; Fukui, T.; Atomi, H.; Imanaka, T.; Miki, K. Crystal Structure of a Novel-Type Archaeal Rubisco with Pentagonal Symmetry. Structure 2001, 9, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Kacar, B.; Hanson-smith, V.; Adam, Z.; Boekelheide, N. Constraining the Timing of the Great Oxidation Event within the Rubisco Phylogenetic Tree. Geobiology 2017, No. May, 628–640. [Google Scholar] [CrossRef]
- Mueller-cajar, O.; Morell, M.; Whitney, S.M. Directed Evolution of Rubisco in Escherichia Coli Reveals a Specificity-Determining Hydrogen Bond in the Form II Enzyme. Biochemistry 2007, 46, 14067–14074. [Google Scholar] [CrossRef]
- Maeda, N.; Kitano, K.; Fukui, T.; Ezaki, S.; Atomi, H.; Miki, K.; Imanaka, T. Ribulose Bisphosphate Carboxylase/Oxygenase from the Hyperthermophilic Archaeon Pyrococcus Kodakaraensis KOD1 Is Composed Solely of Large Subunits and Forms a Pentagonal Structure. J. Mol Biol. 1999, 293, 57–66. [Google Scholar] [CrossRef]
- Nishitani, Y.; Yoshida, S.; Fujihashi, M.; Kitagawa, K.; Doi, T.; Atomi, H.; Imanaka, T.; Miki, K. Structure-Based Catalytic Optimization of a Type III Rubisco from a Hyperthermophile. J. Biol. Chem. 2010, 285, 39339–39347. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Atomi, H.; Imanaka, T. Engineering of a Type III Rubisco from a Hyperthermophilic Archaeon in Order to Enhance Catalytic Performance in Mesophilic Host Cells. Appl. Environ. Microbiol. 2007, 73, 6254–6261. [Google Scholar] [CrossRef] [Green Version]
- Fujihashi, M.; Nishitani, Y.; Kiriyama, T.; Aono, R.; Sato, T.; Takai, T.; Tagashira, K.; Fukuda, W.; Atomi, H.; Imanaka, T.; et al. Mutation Design of a Thermophilic Rubisco Based on Three-Dimensional Structure Enhances Its Activity at Ambient Temperature. Prot. Struct. Funct. Bioinform. 2016, 84, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Dalby, P.A. Coupled Molecular Dynamics Mediate Long- and Short-Range Epistasis between Mutations That Affect Stability and Aggregation Kinetics. Proc. Natl. Acad. Sci. USA 2018, 115, E11043–E11052. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, M.; Szabó, I.; Weetman, S.L.; Sicard, F.; Huber, R.G.; Bond, P.J.; Rosta, E.; Liu, L.N. Molecular Simulations Unravel the Molecular Principles That Mediate Selective Permeability of Carboxysome Shell Protein. Sci. Rep. 2020, 10, 1. [Google Scholar] [CrossRef]
- Tabita, F.R.; Hanson, T.E.; Satagopan, S.; Witte, B.H.; Kreel, N.E. Phylogenetic and Evolutionary Relationships of RubisCO and the RubisCO-like Proteins and the Functional Lessons Provided by Diverse Molecular Forms. R. Soc. 2008, 2629–2640. [Google Scholar] [CrossRef] [PubMed]
- Tabita, F.R.; Satagopan, S.; Hanson, T.E.; Kreel, N.E.; Scott, S.S. Distinct Form I, II, III, and IV Rubisco Proteins from the Three Kingdoms of Life Provide Clues about Rubisco Evolution and Structure/Function Relationships. J. Exp. Botany 2008, 59, 1515–1524. [Google Scholar] [CrossRef]
- Li, H.; Sawaya, M.R.; Tabita, F.R.; Eisenberg, D. Crystal Structure of a RuBisCO-like Protein from the Green Sulfur Bacterium Chlorobium Tepidum. Structure 2005, 13, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Duff, A.P.; Andrews, T.J.; Curmi, P.M.G. The Transition between the Open and Closed States of Rubisco Is Triggered by the Inter-Phosphate Distance of the Bound Bisphosphate. J. Mol. Biol. 2000, 298, 903–916. [Google Scholar] [CrossRef]
- Genkov, T.; Spreitzer, R.J. Highly Conserved Small Subunit Residues Influence Rubisco Large Subunit Catalysis. J. Biol. Chem. 2009, 284, 30105–30112. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissing, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Skjaerven, L.; Yao, X.Q.; Scarabelli, G.; Grant, B.J. Integrating Protein Structural Dynamics and Evolutionary Analysis with Bio3D. BMC Bioinform. 2014, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, B.J.; Skjærven, L.; Yao, X.Q. The Bio3D Packages for Structural Bioinformatics. Prot. Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Structural Bioinformatics Evol and ProDy for Bridging Protein Sequence Evolution and Structural Dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kalenkiewicz, A.; Grant, B.J.; Yang, C.Y. Enrichment of Druggable Conformations from Apo Protein Structures Using Cosolvent-Accelerated Molecular Dynamics. Biology 2015, 4, 344–366. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blow, D. Outline of Crytallography for Biologists, 1st ed.; Oxford University Press: New York, NY, USA, 2002. [Google Scholar]
- Yu-Feng, H. Study of Mining Protein Structural Properties and Its Application; National Taiwan University: Taipei, Taiwan, 2007. [Google Scholar]
- Hanson-Smith, V.; Johnson, A. PhyloBot: A Web Portal for Automated Phylogenetics, Ancestral Sequence Reconstruction, and Exploration of Mutational Trajectories. PLoS Comput. Biol. 2016, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Schmidt, B.; Maskell, D.L. MSAProbs: Multiple Sequence Alignment Based on Pair Hidden Markov Models and Partition Function Posterior Probabilities. Bioinformatics 2010, 26, 1958–1964. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evolution. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Philippe, H. A Bayesian Mixture Model for across-Site Heterogeneities in the Amino-Acid Replacement Process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximim-Likelihood Phylogenies Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2016, 54, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Prot. Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Luthy, R.; Bowie, J.; Eisenberg, D. Assessment of Protein Models with Three-Dimensional Profiles. Nature. 1992, 359, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Bowie, J.; Luthy, R.; Eisenberg, D. A Method to Identify Protein Sequences That Fold into a Known Three-Dimensional Stucture. Science 1991, 253, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; MacKerell, A. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 30, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Ahrari, S.; Khosravi, F.; Osouli, A.; Sakhteman, A.; Nematollahi, A.; Ghasemi, Y.; Savardashtaki, A. MARK4 Protein Can Explore the Active-like Conformations in Its Non-Phosphorylated State. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Salmas, R.E.; Unlu, A.; Yurtsever, M.; Noskov, S.Y.; Durdagi, S. In Silico Investigation of PARP-1 Catalytic Domains in Holo and Apo States for the Design of High-Affinity PARP-1 Inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Guinot, A.D.M. Structural Studies of Different Form I Rubiscos Using Molecular Dynamics Simulations. Doctoral Dissertation, Imperial College London, London, UK, 2016. [Google Scholar] [CrossRef]
- Siqueira, A.S.; Lima, A.R.J.; Dall’Agnol, L.T.; de Azevedo, J.S.N.; da Silva Gonçalves Vianez, J.L.; Gonçalves, E.C. Comparative Modeling and Molecular Dynamics Suggest High Carboxylase Activity of the Cyanobium Sp. CACIAM14 RbcL Protein. J. Mol. Model. 2016, 22, 3. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Yu, H.; Dalby, P.A. A Beginner’s Guide to Molecular Dynamics Simulations and the Identification of Cross-Correlation Networks for Enzyme Engineering. Methods Enzymol. 2020, 643, 15–49. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Grap. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schrodinger, L. The PyMOL Molecular Graphics System, Version 1.3r1. 2010. Available online: https://www.mdpi.com/1422-0067/21/19/7166/htm (accessed on 23 September 2021).
- Martínez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lesgidou, N.; Eliopoulos, E.; Goulielmos, G.; Vlassi, M. Insights on the Alteration of Functionality of a Tyrosine Kinase 2 Variant: A Molecular Dynamics Study. Bioinformatics 2018, 34, i781–i786. [Google Scholar] [CrossRef]
- Hong, L.; Ying, M.; Zheng, C.-J.; Jin, W.-Y.; Liu, W.-S.; Wang, R.-L. Exploring the Effect of D61G Mutation on SHP2 Cause Gain of Function Activity by a Molecular Dynamics Study. J. Biomol. Struct. Dyn. 2018, 36, 3856–3868. [Google Scholar] [CrossRef]
- Rajapaksha, H.; Pandithavidana, D.; Dahanayake, J. Demystifying Chronic Kidney Disease of Unknown Etiology (CKDu): Computational Interaction Analysis of Pesticides and Metabolites with Vital Renal Enzymes. Biomolecules 2021, 11, 261. [Google Scholar] [CrossRef]
- Zalewski, M.; Kmiecik, S.; Kolinski, M. Molecular Dynamics Scoring of Protein–Peptide Models Derived from Coarse-Grained Docking. Moleculaes 2021, 26, 3293. [Google Scholar] [CrossRef]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct Evaluation of Thermal Fluctuations in Proteins Using a Single-Parameter Harmonic Potential. Fold. Design 1997, 2, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.R.; Ma, Y.; Du, S.; Li, W.Y.; Sun, Y.Z.; Zhou, H.; Wang, R.L. Exploring the Reason for Increased Activity of SHP2 Caused by D61Y Mutation through Molecular Dynamics. Comput. Biol. Chem. 2019, 78, 133–143. [Google Scholar] [CrossRef]
- Li, W.Y.; Wei, H.Y.; Sun, Y.Z.; Zhou, H.; Ma, Y.; Wang, R.L. Exploring the Effect of E76K Mutation on SHP2 Cause Gain-of-Function Activity by a Molecular Dynamics Study. J. Cell. Biochem. 2018, 119, 9941–9956. [Google Scholar] [CrossRef]
- Ichiye, T.; Karplus, M. Collective Motions in Proteins: A Covariance Analysis of Atomic Fluctuations in Molecular Dynamics and Normal Mode Simulations. Proteins Struct. Funct. Bioinform. 1991, 11, 205–217. [Google Scholar] [CrossRef]
- Liu, W.S.; Wang, R.R.; Sun, Y.Z.; Li, W.Y.; Li, H.L.; Liu, C.L.; Ma, Y.; Wang, R.L. Exploring the Effect of Inhibitor AKB-9778 on VE-PTP by Molecular Docking and Molecular Dynamics Simulation. J. Cell. Biochem. 2019, 120, 17015–17029. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Z.; Chen, X.B.; Wang, R.R.; Li, W.Y.; Ma, Y. Exploring the Effect of N308D Mutation on Protein Tyrosine Phosphatase-2 Cause Gain-of-Function Activity by a Molecular Dynamics Study. J. Cell. Biochem. 2019, 120, 5949–5961. [Google Scholar] [CrossRef]
- Selvaraj, C.; Omer, A.; Singh, P.; Singh, S. Molecular Insights of Protein Contour Recognition with Ligand Pharmacophoric Sites through Combinatorial Library Design and MD Simulation in Validating HTLV-1 PR Inhibitors. Mol. BioSyst. 2015, 11, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Satagopan, S.; North, J.A.; Arbing, M.A.; Varaljay, V.A.; Haines, S.N.; Wildenthal, J.A.; Byerly, K.M.; Shin, A.; Tabita, F.R. Structural Perturbations of Rhodopseudomonas Palustris Form II RuBisCO Mutant Enzymes That Affect CO2 Fixation. Biochemistry 2019, 58, 3880–3892. [Google Scholar] [CrossRef]
- Ashida, H.; Saito, Y.; Kojima, C.; Kobayashi, K.; Ogasawara, N.; Yokota, A. A Functional Link between RuBisCO-like Protein of Bacillus and Photosynthetic RuBisCO. Science 2003, 302, 86–290. [Google Scholar] [CrossRef] [PubMed]
- Iñiguez, C.; Capó-Bauçà, S.; Niinemets, Ü.; Stoll, H.; Aguiló-Nicolau, P.; Galmés, J. Evolutionary Trends in RuBisCO Kinetics and Their Co-Evolution with CO2 Concentrating Mechanisms. Plant J. 2020, 101, 897–918. [Google Scholar] [CrossRef] [Green Version]
- Poudel, S.; Pike, D.H.; Raanan, H.; Mancini, J.A.; Nanda, V.; Rickaby, R.E.M.; Falkowski, P.G. Biophysical Analysis of the Structural Evolution of Substrate Specificity in RuBisCO. Proc. Natl. Acad. Sci. USA 2020, 117, 30451–30457. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Ashida, H.; Sakiyama, T.; de Marsac, N.T.; Danchin, A.; Sekowska, A.; Yokota, A. Structural and Functional Similarities between a Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase (RuBisCO)-like Protein from Bacillus Subtilis and Photosynthetic RuBisCO. J. Biol. Chem. 2009, 284, 13256–13264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashida, H.; Saito, Y.; Nakano, T.; Tandeau De Marsac, N.; Sekowska, A.; Danchin, A.; Yokota, A. RuBisCO-like Proteins as the Enolase Enzyme in the Methionine Salvage Pathway: Functional and Evolutionary Relationships between RuBisCO-like Proteins and Photosynthetic RuBisCO. J. Exp. Botany 2008, 59, 1543–1554. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, H.; Yamamoto, H.; Shibata, N.; Inoue, T.; Okada, S.; Miyake, C.; Yokota, A.; Yasushi, K. Crystal Structure of Carboxylase Reaction-Oriented Ribulose 1,5- Bisphosphate Carboxylase/Oxygenase from a Thermophilic Red Alga, Galdieria Partita. J. Biol. Chem. 1999, 274, 15655–15661. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Stessman, D.; Spalding, M. The CO2 Concentrating Mechanism and Photosynthetic Carbon Assimilation in Limiting CO2: How Chlamydomonas Works against the Gradient. Plant J. 2015, 82, 429–448. [Google Scholar] [CrossRef]
- Banda, D.M.; Pereira, J.H.; Liu, A.K.; Orr, D.J.; Hammel, M.; He, C.; Parry, M.A.J.; Carmo-Silva, E.; Adams, P.D.; Banfield, J.F.; et al. Novel Bacterial Clade Reveals Origin of Form I Rubisco. Nat. Plants 2020, 6, 1158–1166. [Google Scholar] [CrossRef]
- Alonso, H.; Blayney, M.J.; Beck, J.L.; Whitney, S.M. Substrate-Induced Assembly of Methanococcoides Burtonii D-Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase Dimers into Decamers. J. Biol. Chem. 2009, 284, 33876–33882. [Google Scholar] [CrossRef] [Green Version]
- Gunn, L.H.; Valegard, K.; Andersson, I. A Unique Structural Domain in Methanococcoides Burtonii Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase (Rubisco) Acts as a Small Subunit Mimic. J. Biol. Chem. 2017, 292, 6838–6850. [Google Scholar] [CrossRef] [Green Version]
- Schreuder, H.A.; Knight, S.; Curmi, P.M.G.; Andersson, I.; Cascio, D.; Branden, C.I.; Eisenberg, D. Formation of the Active Site of Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase by a Disorder-Order Transition from the Unactivated to the Activated Form. Proc. Natl. Acad. Sci. USA 1993, 90, 9968–9972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seno, Y.; Go, N. Deoxymyoglobin Studied by the Conformational Normal Mode Analysis. I. Dynamics of Globin and the Heme-Globin Interaction. J. Mol. Biol. 1990, 216, 95–109. [Google Scholar] [CrossRef]
- Levitt, M.; Sander, C.; Stern, P.S. Protein Normal-Mode Dynamics: Trypsin Inhibitor, Crambin, Ribonuclease and Lysozyme. J. Mol. Biol. 1985, 181, 423–447. [Google Scholar] [CrossRef]
- Schloss, J.V. Comparative Affinities of the Epimeric Reaction-Intermediate Analogs 2- and 4-Carboxy-D-Arabinitol 1,5-Bisphosphate for Spinach Ribulose 1,5-Bisphosphate Carboxylase. J. Biol. Chem. 1988, 263, 4145–4150. [Google Scholar] [CrossRef]
- Ng, J.; Guo, Z.; Mueller-Cajar, O. Rubisco Activase Requires Residues in the Large Subunit N Terminus to Remodel Inhibited Plant Rubisco. J. Biol. Chem. 2020, 295, 16427–16435. [Google Scholar] [CrossRef]
- Watanabe, H.; Enomoto, T.; Tanaka, S. Ab Initio Study of Molecular Interactions in Higher Plant and Galdieria Partita Rubiscos with the Fragment Molecular Orbital Method. Biochem. Biophys. Res. Commun. 2007, 361, 367–372. [Google Scholar] [CrossRef]
- Satagopan, S.; Chan, S.; Perry, L.J.; Tabita, F.R. Structure-Function Studies with the Unique Hexameric Form II Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase (Rubisco) from Rhodopseudomonas Palustris. J. Biol. Chem. 2014, 289, 21433–21450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller-Cajar, O. The Diverse AAA+ Machines That Repair Inhibited Rubisco Active Sites. Front. Mol. Biosci. 2017, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Taylor, T.; Andersson, I. Structural Transitions during Activation and Ligand Binding in Hexadecameric Rubisco Inferred from the Crystal Structure of the Activated Unliganded Spinach Enzyme. Nat. Struct. Biol. 1996, 3, 95–101. [Google Scholar] [CrossRef]
- Atomi, H.; Fukui, T.; Kanai, T.; Morikawa, M.; Imanaka, T. Description of Thermococcus Kodakaraensis Sp. Nov., a Well Studied Hyperthermophilic Archaeon Previously Reported as Pyrococcus Sp. KOD1. Archaea 2004, 1, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.; Choi, S. Structure-Activity Relationship in TLR4 Mutations: Atomistic Molecular Dynamics Simulations and Residue Interaction Network Analysis. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Form | Protein | RMSD | Region ≥0.3 of RMSF | Sequence | Structures |
|---|---|---|---|---|---|
| I | 1WDDWT | 0.52 ± 0.004 | 68–76 | TVWTDGLTS | Loop connecting αB and βC |
| 4RUBWT | 0.36 ± 0.002 | 64–75; 125; 209–211 | GTWTTVWTDGLT; F; QPF | Loop connecting αB and βC; α0; Loop connecting β2 and α2 | |
| 5IU0WT | 0.46 ± 0.003 | 22; 66–75 | L; WTTVWTDGLT | N-terminal tail; Loop connecting αB and βC | |
| 1IWAWT | 0.44 ± 0.003 | 55–56; 73–86; 482 | PG; WTVVWTDLLTAA; T | βB; Loop connecting αB and βC; C-terminal tail | |
| 1GK8WT | 0.34 ± 0.002 | 69–75 | VWTDGLT | Loop connecting αB and βC | |
| 6FTLWT | 0.37 ± 0.002 | 71–80; 211–212 | TVVWTDLLTA; NS | Loop connecting αB and βC; Loop connecting β2 and α2 | |
| II | 4LF1WT | 0.24 ± 0.001 | 53–63; 201–202 | GTNVEVSTTDD; VF | Loop connecting αB and βC; Loop connecting β2 and α2 |
| 5HANS59F | 0.28 ± 0.002 | 56–65 | VEVFTTDDFT | Loop connecting αB and βC | |
| 5HJXA47V | 0.34 ± 0.002 | 54–63; 330–331 | TNVEVSTTDD; KM | Loop connecting αB and βC; Loop 6 | |
| III | 3A12WT | 0.18 ± 0.001 | 58–59; 286 | LY; A | Loop connecting αB and βC; αF |
| 3KDOSP6 | 0.28 ± 0.002 | 55–62; 347 | WTTLYPWY; N | Loop connecting αB and βC; Loop connecting α6 and β7 | |
| 3WQPT289D | 0.22 ± 0.001 | 57–63; 322 | TLYPWYE; K | Loop connecting αB and βC; Loop 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camel, V.; Zolla, G. An Insight of RuBisCO Evolution through a Multilevel Approach. Biomolecules 2021, 11, 1761. https://doi.org/10.3390/biom11121761
Camel V, Zolla G. An Insight of RuBisCO Evolution through a Multilevel Approach. Biomolecules. 2021; 11(12):1761. https://doi.org/10.3390/biom11121761
Chicago/Turabian StyleCamel, Vladimir, and Gaston Zolla. 2021. "An Insight of RuBisCO Evolution through a Multilevel Approach" Biomolecules 11, no. 12: 1761. https://doi.org/10.3390/biom11121761
APA StyleCamel, V., & Zolla, G. (2021). An Insight of RuBisCO Evolution through a Multilevel Approach. Biomolecules, 11(12), 1761. https://doi.org/10.3390/biom11121761