
Transcription Factors Active in the Anterior Blastema of Schmidtea mediterranea

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Planarian Husbandry
2.2. Preparation of Blastema Samples
2.3. Flow Cytometry Analysis and FACS
2.4. Irradiation and RNAi
2.5. RNA Extraction and cDNA Synthesis
2.6. RT-PCR and RT-qPCR
2.7. RNA-Seq
2.7.1. cDNA Library Preparation
2.7.2. PCR, Emulsion PCR and RNA-Seq Run
2.7.3. RNA-Seq Data Primary Analysis
2.8. RNA-Seq Analytical Pipeline
2.9. Whole Mount In Situ Hybridization, Fluorescent In Situ Hybridization and Immunohistochemistry
2.10. Gene Specific Knock-Down by dsRNA Injection
2.11. Statistical Analysis
3. Results
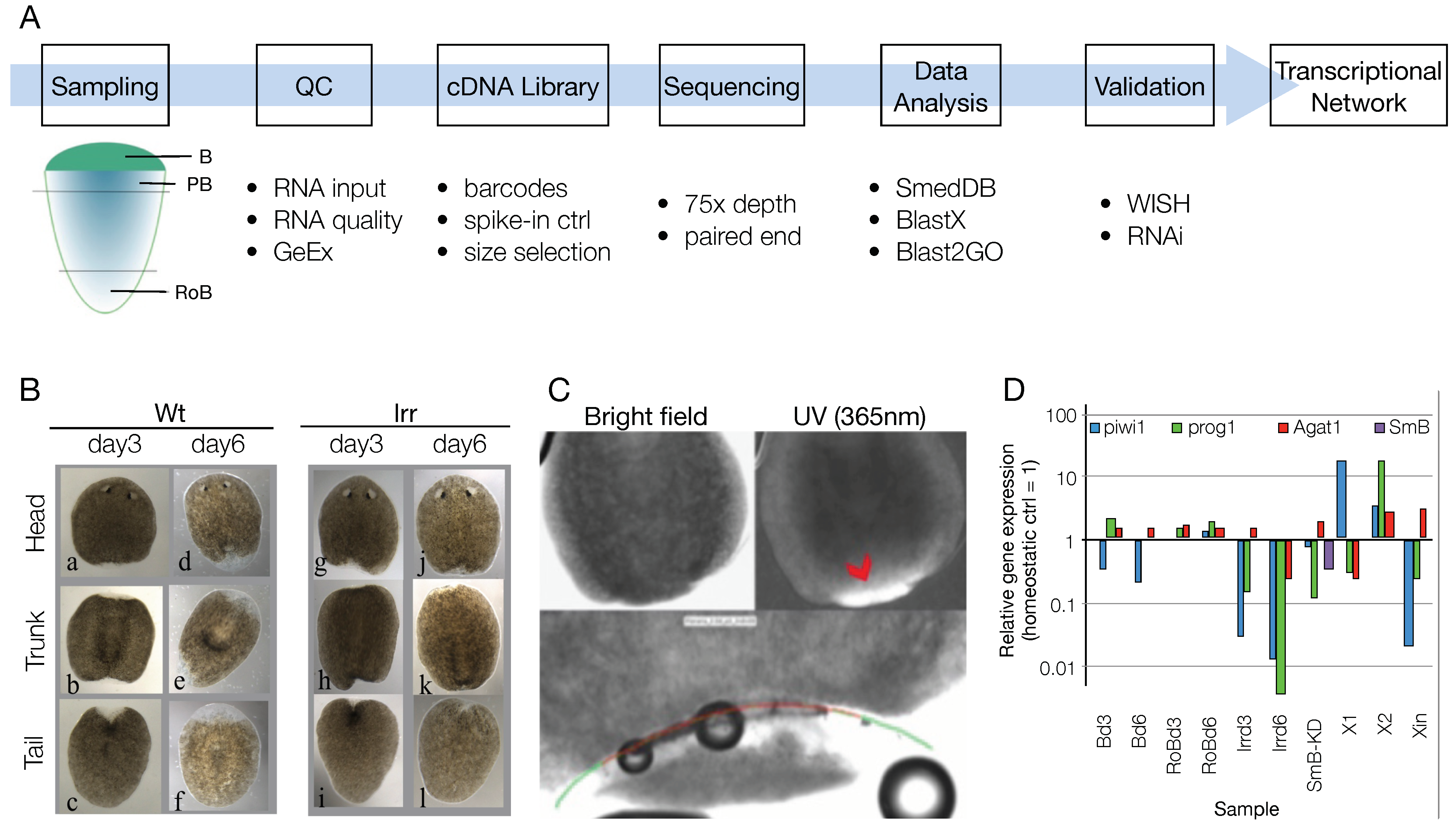
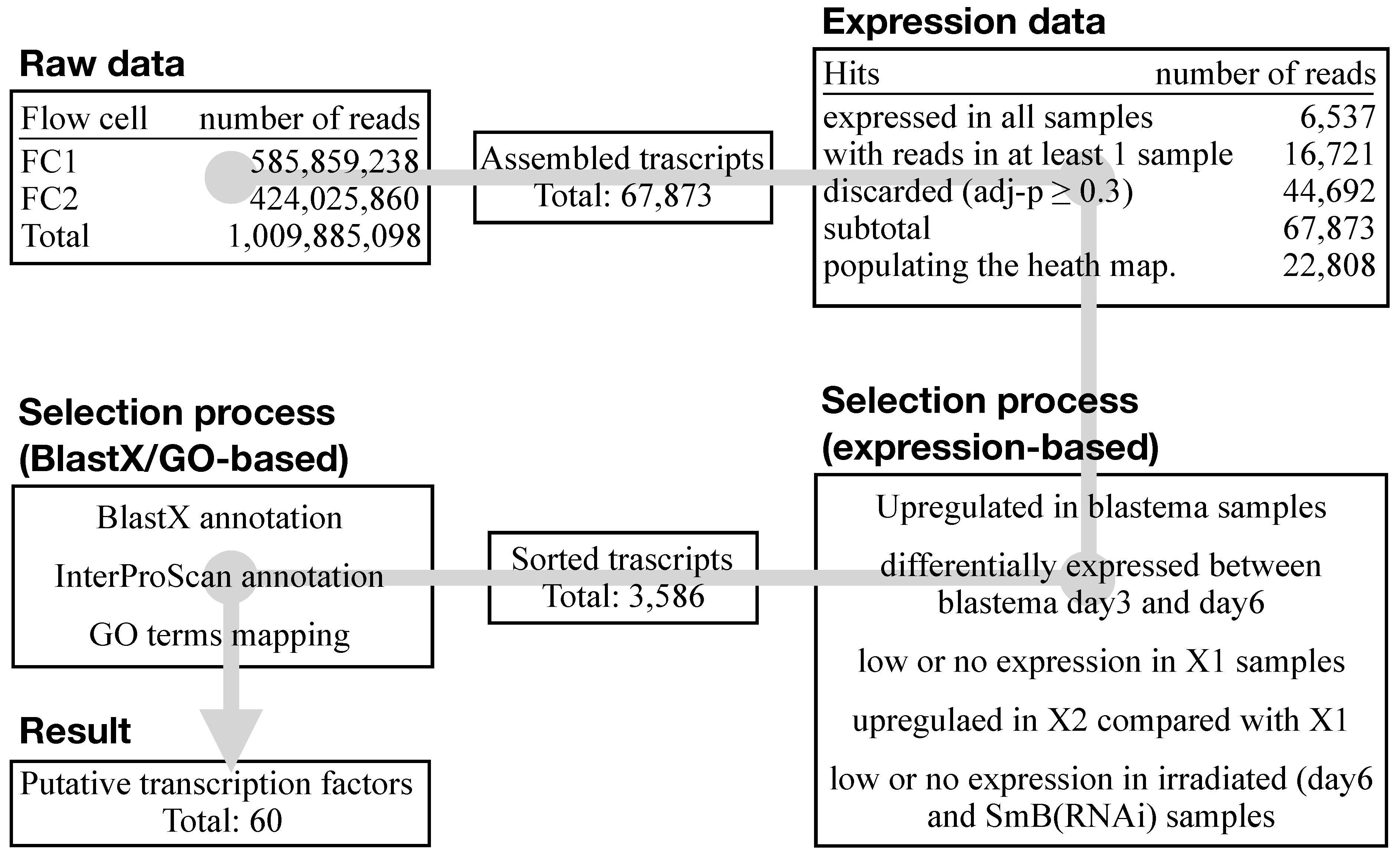
3.1. Sample List Definition and Pre-Run Quality Check
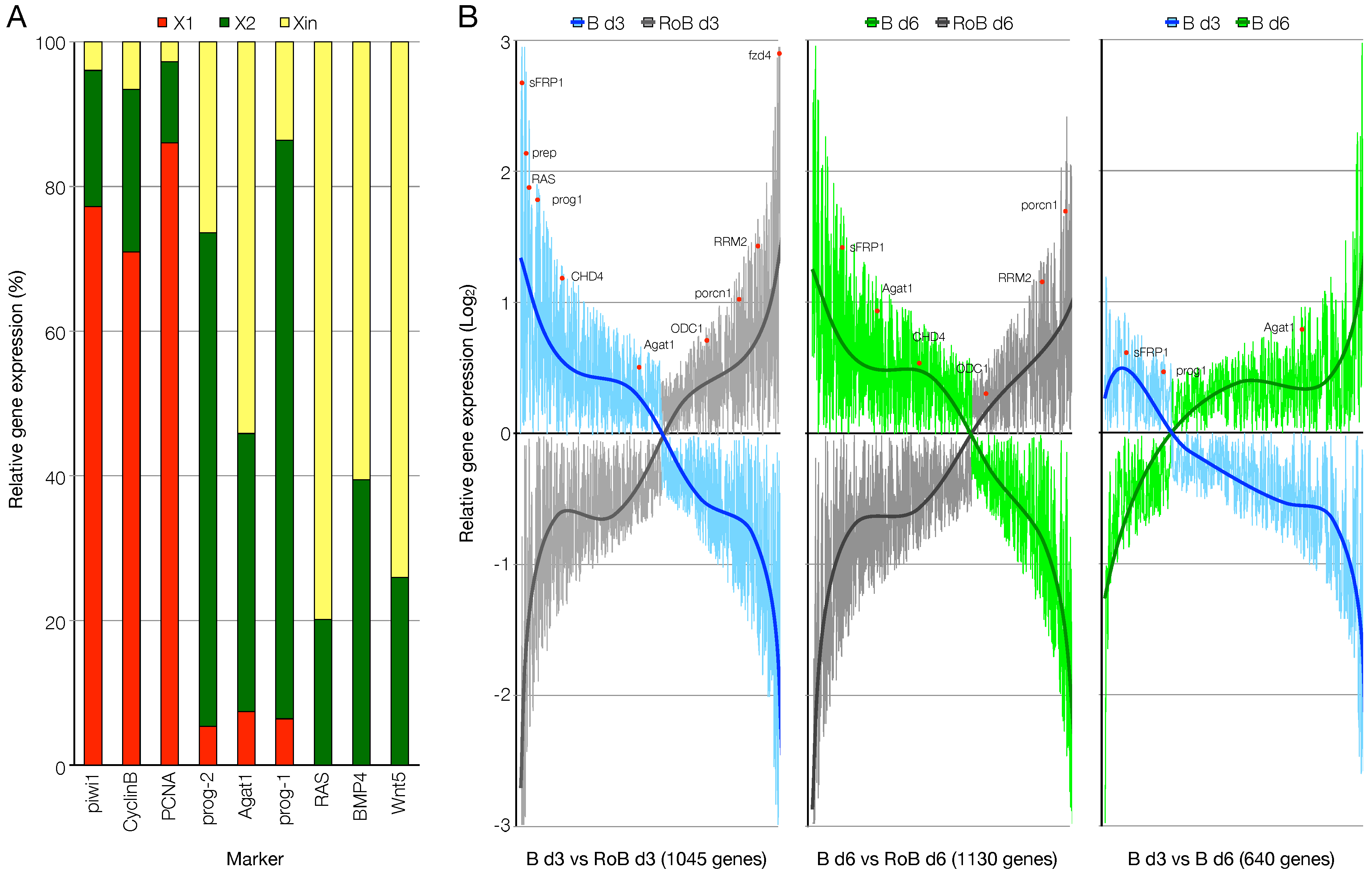
3.2. RNA-Seq Data Quality Check Showed High Quality Scores and Confirmed the Upregulation of Known Markers of Cell Commitment and Differentiation in the Blastema Samples
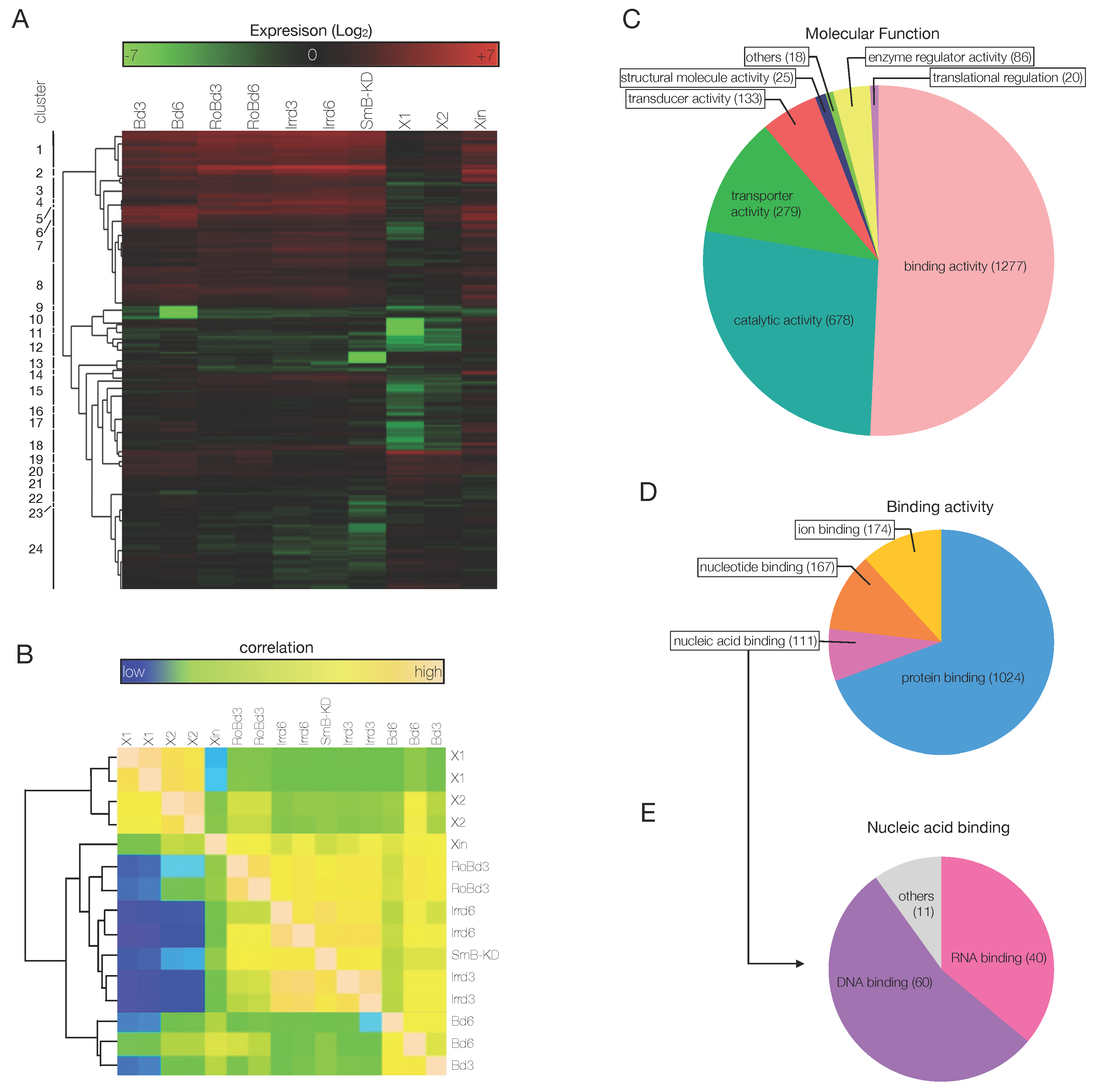
3.3. An Ad Hoc Analysis Pipeline Sorted 65 Putative Transcription Factors Active in the Blastema
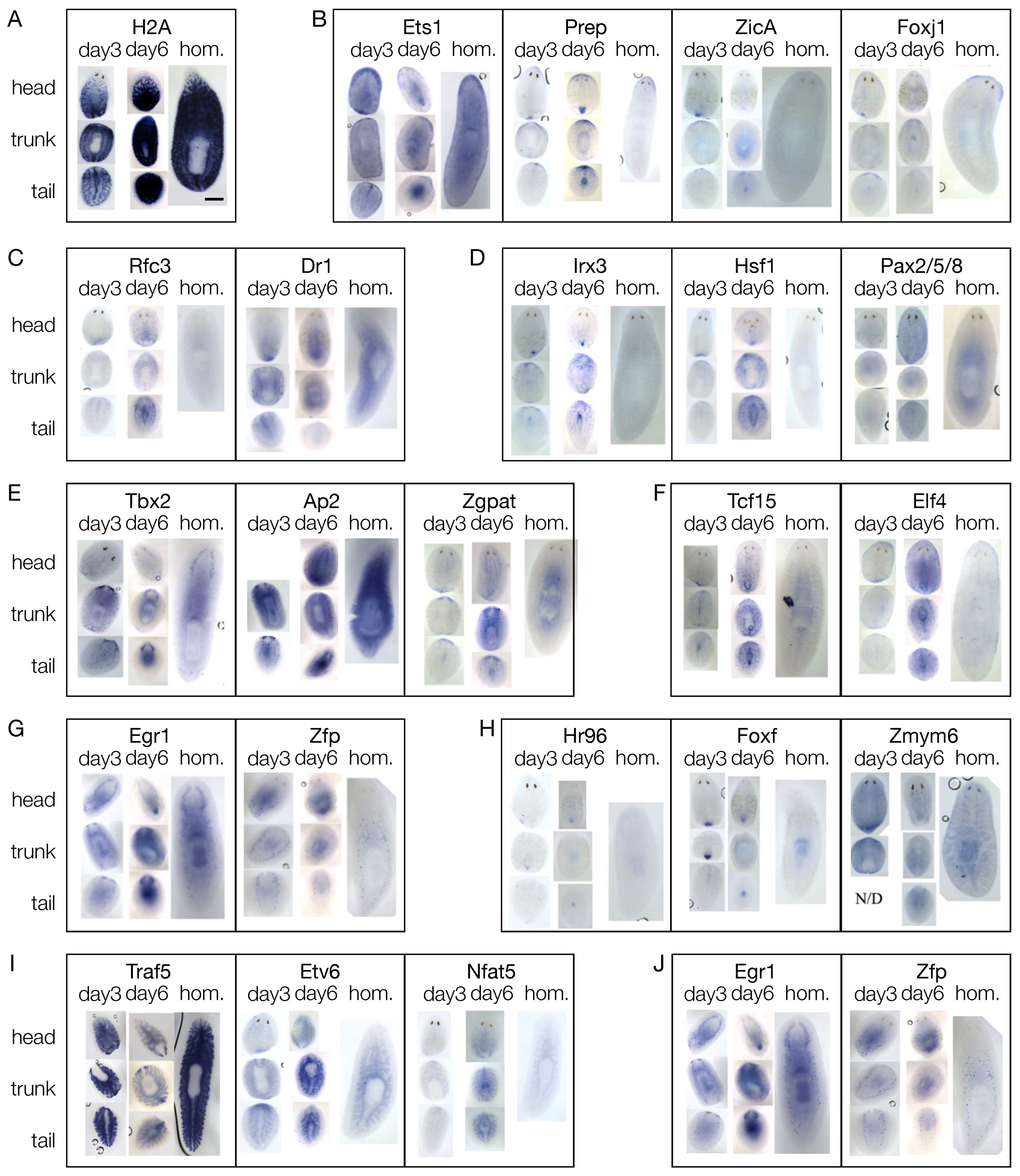
3.4. The mRNAs of 44 Shortlisted Transcription Factors were Found Enriched in the Regenerating Blastema
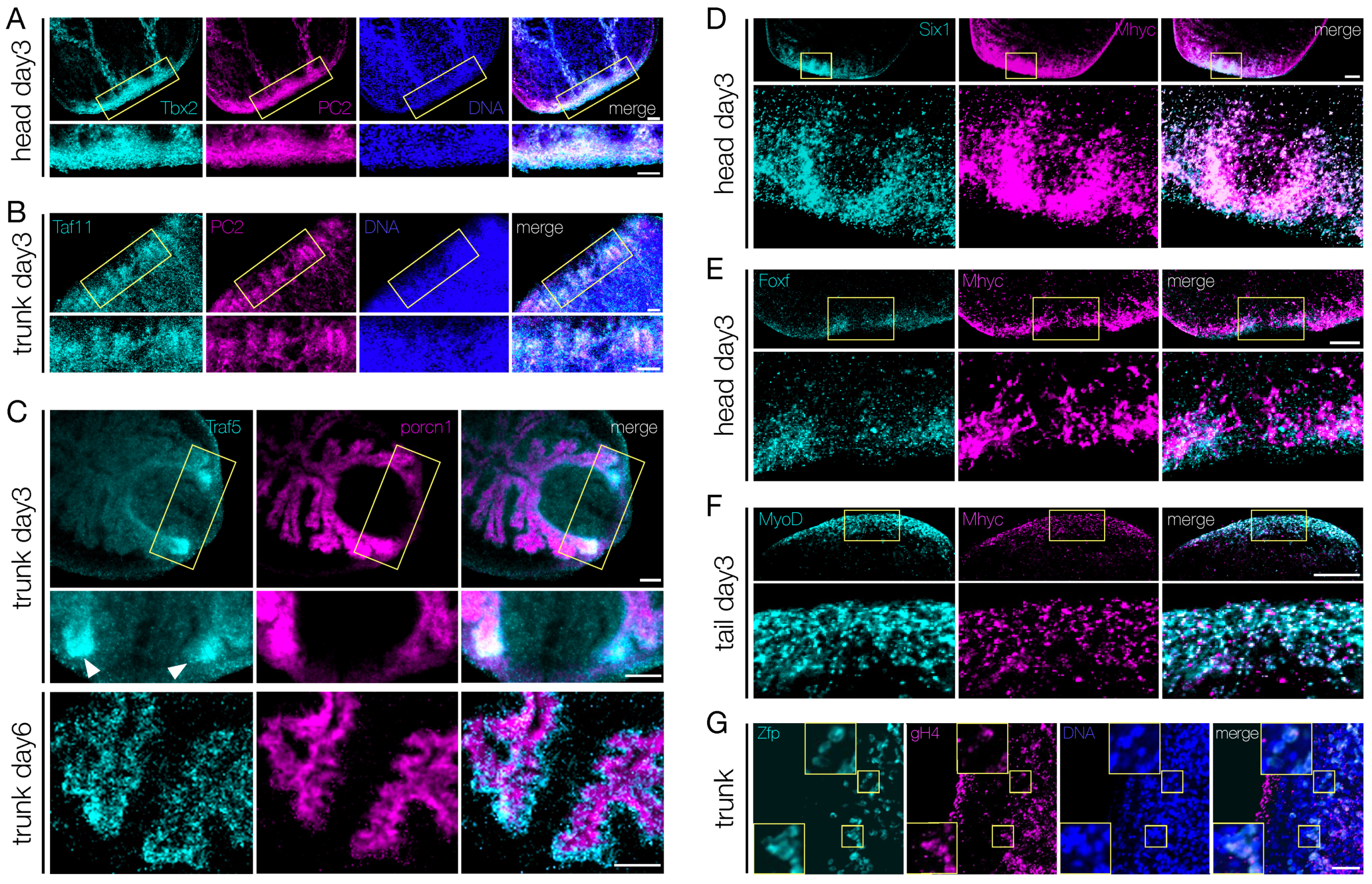
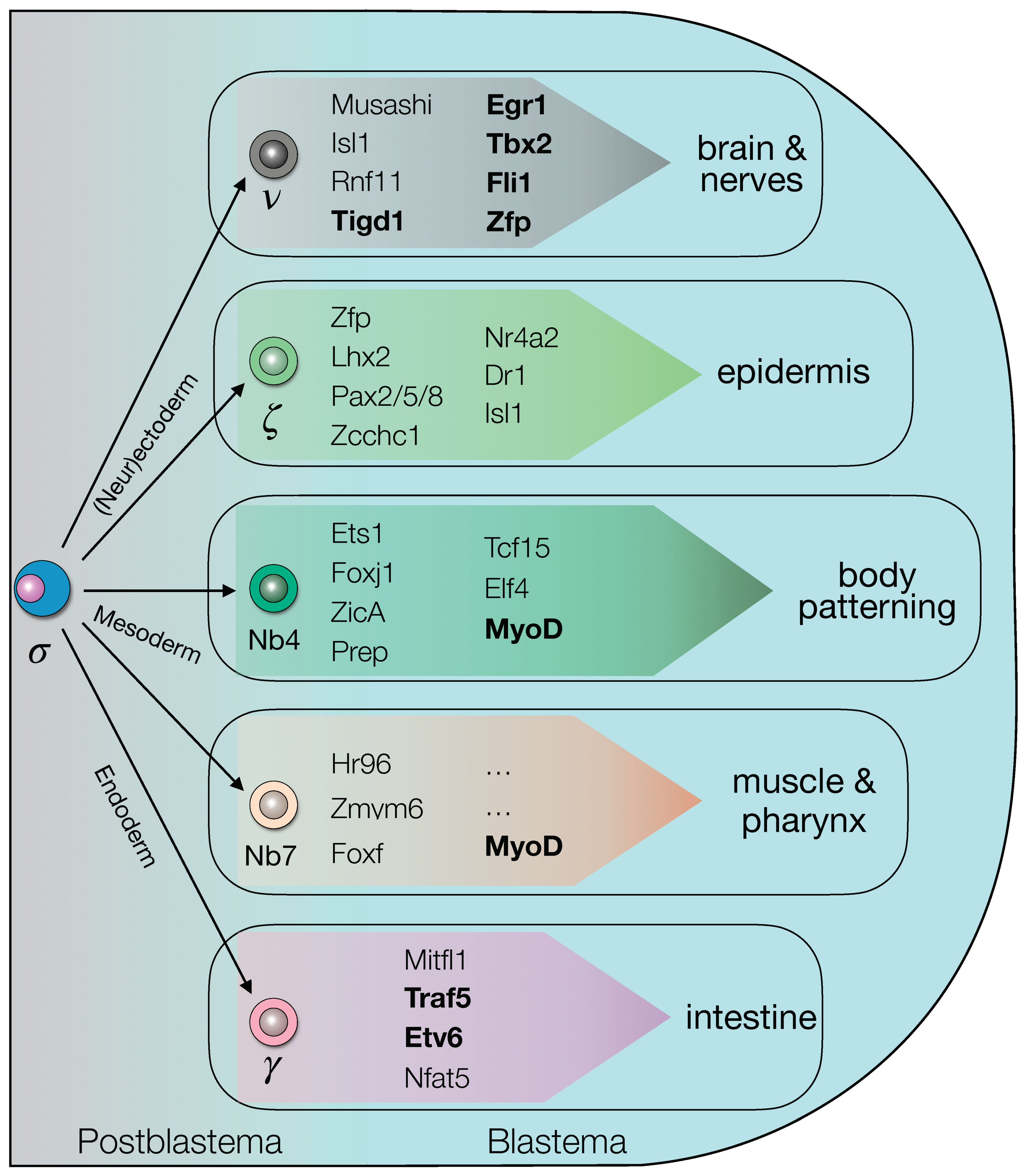
3.5. Shortlisted Blastema Transcription Factors Play a Role in Regeneration
4. Discussion
4.1. The Sampling Strategy and the Ad Hoc Defined Analytical Pipeline Effectively Shortlisted Blastema TFs
4.2. The Blastema TFs Are Active in the Specification and Differentiation of the Regenerating Structures, including the Cells Responsible for Body Patterning and Positional Instruction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cebria, F. Regenerating the central nervous system: How easy for planarians! Dev. Genes Evol. 2007, 217, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Agata, K.; Umesono, Y. Brain regeneration from pluripotent stem cells in planarian. Philos. Trans. R Soc. Lond. B Biol. Sci. 2008, 363, 2071–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, N.; Hayashi, T.; Fukumura, R.; Fujii, J.; Kudome-Takamatsu, T.; Nishimura, O.; Sano, S.; Son, F.; Suzuki, N.; Araki, R.; et al. Comprehensive gene expression analyses in pluripotent stem cells of a planarian, Dugesia japonica. Int. J. Dev. Biol. 2012, 56, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Currie, K.W.; Pearson, B.J. Transcription factors lhx1/5-1 and pitx are required for the maintenance and regeneration of serotonergic neurons in planarians. Development 2013, 140, 3577–3588. [Google Scholar] [CrossRef] [Green Version]
- Marz, M.; Seebeck, F.; Bartscherer, K. A Pitx transcription factor controls the establishment and maintenance of the serotonergic lineage in planarians. Development 2013, 140, 4499–4509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowles, M.W.; Omuro, K.C.; Stanley, B.N.; Quintanilla, C.G.; Zayas, R.M. COE loss-of-function analysis reveals a genetic program underlying maintenance and regeneration of the nervous system in planarians. PLoS Genet. 2014, 10, e1004746. [Google Scholar] [CrossRef] [Green Version]
- Currie, K.W.; Molinaro, A.M.; Pearson, B.J. Neuronal sources of hedgehog modulate neurogenesis in the adult planarian brain. eLife 2016, 5, e19735. [Google Scholar] [CrossRef] [Green Version]
- Roberts-Galbraith, R.H.; Brubacher, J.L.; Newmark, P.A. A functional genomics screen in planarians reveals regulators of whole-brain regeneration. eLife 2016, 5, e17002. [Google Scholar] [CrossRef]
- Wang, I.E.; Lapan, S.W.; Scimone, M.L.; Clandinin, T.R.; Reddien, P.W. Hedgehog signaling regulates gene expression in planarian glia. eLife 2016, 5, e16996. [Google Scholar] [CrossRef] [PubMed]
- Karge, A.; Bonar, N.A.; Wood, S.; Petersen, C.P. tec-1 kinase negatively regulates regenerative neurogenesis in planarians. eLife 2020, 9, e47293. [Google Scholar] [CrossRef] [PubMed]
- Lapan, S.W.; Reddien, P.W. dlx and sp6-9 Control optic cup regeneration in a prototypic eye. PLoS Genet. 2011, 7, e1002226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Agata, K. Optic chiasm formation in planarian I: Cooperative netrin- and robo-mediated signals are required for the early stage of optic chiasm formation. Dev. Growth Differ. 2011, 53, 300–311. [Google Scholar] [CrossRef]
- Gonzalez-Sastre, A.; Molina, M.D.; Salo, E. Inhibitory Smads and bone morphogenetic protein (BMP) modulate anterior photoreceptor cell number during planarian eye regeneration. Int. J. Dev. Biol. 2012, 56, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapan, S.W.; Reddien, P.W. Transcriptome analysis of the planarian eye identifies ovo as a specific regulator of eye regeneration. Cell Rep. 2012, 2, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.; Wang, I.E.; Reddien, P.W. pbx is required for pole and eye regeneration in planarians. Development 2013, 140, 719–729. [Google Scholar] [CrossRef] [Green Version]
- LoCascio, S.A.; Lapan, S.W.; Reddien, P.W. Eye Absence Does Not Regulate Planarian Stem Cells during Eye Regeneration. Dev. Cell 2017, 40, 381–391.e383. [Google Scholar] [CrossRef] [Green Version]
- Atabay, K.D.; LoCascio, S.A.; de Hoog, T.; Reddien, P.W. Self-organization and progenitor targeting generate stable patterns in planarian regeneration. Science 2018, 360, 404–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emili, E.; Esteve Pallares, M.; Romero, R.; Cebria, F. Smed-egfr-4 is required for planarian eye regeneration. Int. J. Dev. Biol. 2019, 63, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A.; Mukundan, N.; Sowndarya, S.; Dubey, V.K.; Babu, R.; Lakshmanan, V.; Rangiah, K.; Panicker, M.M.; Palakodeti, D.; Subramanian, S.P.; et al. Serotonin is essential for eye regeneration in planaria Schmidtea mediterranea. FEBS Lett. 2019, 593, 3198–3209. [Google Scholar] [CrossRef]
- Scimone, M.L.; Atabay, K.D.; Fincher, C.T.; Bonneau, A.R.; Li, D.J.; Reddien, P.W. Muscle and neuronal guidepost-like cells facilitate planarian visual system regeneration. Science 2020, 368, 3203. [Google Scholar] [CrossRef]
- Reddien, P.W. Principles of regeneration revealed by the planarian eye. Curr. Opin. Cell Biol. 2021, 73, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.C.; Vu, H.T.; Sanchez Alvarado, A. The maintenance and regeneration of the planarian excretory system are regulated by EGFR signaling. Development 2011, 138, 3769–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scimone, M.L.; Srivastava, M.; Bell, G.W.; Reddien, P.W. A regulatory program for excretory system regeneration in planarians. Development 2011, 138, 4387–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thi-Kim Vu, H.; Rink, J.C.; McKinney, S.A.; McClain, M.; Lakshmanaperumal, N.; Alexander, R.; Sanchez Alvarado, A. Stem cells and fluid flow drive cyst formation in an invertebrate excretory organ. eLife 2015, 4, e07405. [Google Scholar] [CrossRef] [PubMed]
- Forsthoefel, D.J.; Park, A.E.; Newmark, P.A. Stem cell-based growth, regeneration, and remodeling of the planarian intestine. Dev. Biol. 2011, 356, 445–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, N.M.; Oviedo, N.J.; Sage, J. Essential role for the planarian intestinal GATA transcription factor in stem cells and regeneration. Dev. Biol. 2016, 418, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Sastre, A.; De Sousa, N.; Adell, T.; Salo, E. The pioneer factor Smed-gata456-1 is required for gut cell differentiation and maintenance in planarians. Int. J. Dev. Biol. 2017, 61, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsthoefel, D.J.; Cejda, N.I.; Khan, U.W.; Newmark, P.A. Cell-type diversity and regionalized gene expression in the planarian intestine. eLife 2020, 9, e52613. [Google Scholar] [CrossRef] [PubMed]
- Witchley, J.N.; Mayer, M.; Wagner, D.E.; Owen, J.H.; Reddien, P.W. Muscle cells provide instructions for planarian regeneration. Cell Rep. 2013, 4, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Cebria, F. Planarian Body-Wall Muscle: Regeneration and Function beyond a Simple Skeletal Support. Front. Cell Dev. Biol. 2016, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Adler, C.E.; Sanchez Alvarado, A. PHRED-1 is a divergent neurexin-1 homolog that organizes muscle fibers and patterns organs during regeneration. Dev. Biol. 2017, 427, 165–175. [Google Scholar] [CrossRef]
- Moritz, S.; Stockle, F.; Ortmeier, C.; Schmitz, H.; Rodriguez-Esteban, G.; Key, G.; Gentile, L. Heterogeneity of planarian stem cells in the S/G2/M phase. Int. J. Dev. Biol. 2012, 56, 117–125. [Google Scholar] [CrossRef] [Green Version]
- van Wolfswinkel, J.C.; Wagner, D.E.; Reddien, P.W. Single-cell analysis reveals functionally distinct classes within the planarian stem cell compartment. Cell Stem Cell 2014, 15, 326–339. [Google Scholar] [CrossRef] [Green Version]
- Molinaro, A.M.; Pearson, B.J. In silico lineage tracing through single cell transcriptomics identifies a neural stem cell population in planarians. Genome Biol. 2016, 17, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurtzel, O.; Oderberg, I.M.; Reddien, P.W. Planarian Epidermal Stem Cells Respond to Positional Cues to Promote Cell-Type Diversity. Dev. Cell 2017, 40, 491–504.e495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, A.; Li, H.; Guo, L.; Gao, X.; McKinney, S.; Wang, Y.; Yu, Z.; Park, J.; Semerad, C.; Ross, E.; et al. Prospectively Isolated Tetraspanin(+) Neoblasts Are Adult Pluripotent Stem Cells Underlying Planaria Regeneration. Cell 2018, 173, 1593–1608.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, K.; Xu, H.; Xiong, Y.Z.; Zhao, Y.; Gao, C.; Seidel, C.W.; Pan, X.; Ying, Y.; Lei, K. Canonical and early lineage-specific stem cell types identified in planarian SirNeoblasts. Cell Regen. 2021, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Raz, A.A.; Wurtzel, O.; Reddien, P.W. Planarian stem cells specify fate yet retain potency during the cell cycle. Cell Stem Cell 2021, 28, 1307–1322.e1305. [Google Scholar] [CrossRef] [PubMed]
- Felix, D.A.; Aboobaker, A.A. The TALE class homeobox gene Smed-prep defines the anterior compartment for head regeneration. PLoS Genet. 2010, 6, e1000915. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Motoishi, M.; Yazawa, S.; Itomi, K.; Tanegashima, C.; Nishimura, O.; Agata, K.; Tarui, H. A LIM-homeobox gene is required for differentiation of Wnt-expressing cells at the posterior end of the planarian body. Development 2011, 138, 3679–3688. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, S.; Hayashi, T.; Tarui, H.; Nishimura, O.; Nishimura, K.; Shibata, N.; Sakamoto, H.; Agata, K. Expression and functional analysis of musashi-like genes in planarian CNS regeneration. Mech. Dev. 2008, 125, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.E.; Wang, I.E.; Reddien, P.W. Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration. Science 2011, 332, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Esteban, G.; Gonzalez-Sastre, A.; Rojo-Laguna, J.I.; Salo, E.; Abril, J.F. Digital gene expression approach over multiple RNA-Seq data sets to detect neoblast transcriptional changes in Schmidtea mediterranea. BMC Genom. 2015, 16, 361. [Google Scholar] [CrossRef] [Green Version]
- Fincher, C.T.; Wurtzel, O.; de Hoog, T.; Kravarik, K.M.; Reddien, P.W. Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science 2018, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenemoser, D.; Lapan, S.W.; Wilkinson, A.W.; Bell, G.W.; Reddien, P.W. A molecular wound response program associated with regeneration initiation in planarians. Genes Dev. 2012, 26, 988–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abril, J.F.; Cebria, F.; Rodriguez-Esteban, G.; Horn, T.; Fraguas, S.; Calvo, B.; Bartscherer, K.; Salo, E. Smed454 dataset: Unravelling the transcriptome of Schmidtea mediterranea. BMC Genom. 2010, 11, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blythe, M.J.; Kao, D.; Malla, S.; Rowsell, J.; Wilson, R.; Evans, D.; Jowett, J.; Hall, A.; Lemay, V.; Lam, S.; et al. A dual platform approach to transcript discovery for the planarian Schmidtea mediterranea to establish RNAseq for stem cell and regeneration biology. PLoS ONE 2010, 5, e15617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamidi, C.; Wang, Y.; Gruen, D.; Mastrobuoni, G.; You, X.; Tolle, D.; Dodt, M.; Mackowiak, S.D.; Gogol-Doering, A.; Oenal, P.; et al. De novo assembly and validation of planaria transcriptome by massive parallel sequencing and shotgun proteomics. Genome Res. 2011, 21, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.F.; Fang, H.M.; Tian, Q.N.; Bao, Z.X.; Lu, P.; Zhao, J.M.; Mai, J.; Zhu, Z.Y.; Shu, L.L.; Zhao, L.; et al. Transcriptome profiling and digital gene expression by deep-sequencing in normal/regenerative tissues of planarian Dugesia japonica. Genomics 2011, 97, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandmann, T.; Vogg, M.C.; Owlarn, S.; Boutros, M.; Bartscherer, K. The head-regeneration transcriptome of the planarian Schmidtea mediterranea. Genome Biol. 2011, 12, R76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloni, M. Global irradiation effects, stem cell genes and rare transcripts in the planarian transcriptome. Int. J. Dev. Biol. 2012, 56, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbe, R.M.; Irimia, M.; Currie, K.W.; Lin, A.; Zhu, S.J.; Brown, D.D.; Ross, E.J.; Voisin, V.; Bader, G.D.; Blencowe, B.J.; et al. A comparative transcriptomic analysis reveals conserved features of stem cell pluripotency in planarians and mammals. Stem Cells 2012, 30, 1734–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, O.; Hirao, Y.; Tarui, H.; Agata, K. Comparative transcriptome analysis between planarian Dugesia japonica and other platyhelminth species. BMC Genom. 2012, 13, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onal, P.; Grun, D.; Adamidi, C.; Rybak, A.; Solana, J.; Mastrobuoni, G.; Wang, Y.; Rahn, H.P.; Chen, W.; Kempa, S.; et al. Gene expression of pluripotency determinants is conserved between mammalian and planarian stem cells. EMBO J. 2012, 31, 2755–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resch, A.M.; Palakodeti, D.; Lu, Y.C.; Horowitz, M.; Graveley, B.R. Transcriptome analysis reveals strain-specific and conserved stemness genes in Schmidtea mediterranea. PLoS ONE 2012, 7, e34447. [Google Scholar] [CrossRef]
- Solana, J.; Kao, D.; Mihaylova, Y.; Jaber-Hijazi, F.; Malla, S.; Wilson, R.; Aboobaker, A. Defining the molecular profile of planarian pluripotent stem cells using a combinatorial RNAseq, RNA interference and irradiation approach. Genome Biol. 2012, 13, R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, D.; Felix, D.; Aboobaker, A. The planarian regeneration transcriptome reveals a shared but temporally shifted regulatory program between opposing head and tail scenarios. BMC Genom. 2013, 14, 797. [Google Scholar] [CrossRef] [Green Version]
- Sikes, J.M.; Newmark, P.A. Restoration of anterior regeneration in a planarian with limited regenerative ability. Nature 2013, 500, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Scimone, M.L.; Kravarik, K.M.; Lapan, S.W.; Reddien, P.W. Neoblast specialization in regeneration of the planarian Schmidtea mediterranea. Stem Cell Rep. 2014, 3, 339–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.; Mazza-Curll, K.L.; van Wolfswinkel, J.C.; Reddien, P.W. Whole-body acoel regeneration is controlled by Wnt and Bmp-Admp signaling. Curr. Biol. 2014, 24, 1107–1113. [Google Scholar] [CrossRef] [Green Version]
- Vogg, M.C.; Owlarn, S.; Perez Rico, Y.A.; Xie, J.; Suzuki, Y.; Gentile, L.; Wu, W.; Bartscherer, K. Stem cell-dependent formation of a functional anterior regeneration pole in planarians requires Zic and Forkhead transcription factors. Dev. Biol. 2014, 390, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, H.; Marz, M.; Vogg, M.C.; Eccles, D.; Grifol-Boldu, L.; Wehner, D.; Owlarn, S.; Adell, T.; Weidinger, G.; Bartscherer, K. Beta-catenin-dependent control of positional information along the AP body axis in planarians involves a teashirt family member. Cell Rep. 2015, 10, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, N.J.; Agbedanu, P.N.; Kimber, M.J.; Ribeiro, P.; Day, T.A.; Zamanian, M. Functional analysis of Girardia tigrina transcriptome seeds pipeline for anthelmintic target discovery. Parasites Vectors 2015, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Pang, Q.; Gao, L.; Hu, W.; An, Y.; Deng, H.; Zhang, Y.; Sun, X.; Zhu, G.; Liu, B.; Zhao, B. De Novo Transcriptome Analysis Provides Insights into Immune Related Genes and the RIG-I-Like Receptor Signaling Pathway in the Freshwater Planarian (Dugesia japonica). PLoS ONE 2016, 11, e0151597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raz, A.A.; Srivastava, M.; Salvamoser, R.; Reddien, P.W. Acoel regeneration mechanisms indicate an ancient role for muscle in regenerative patterning. Nat. Commun. 2017, 8, 1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, Y.; Abnave, P.; Kao, D.; Hughes, S.; Lai, A.; Jaber-Hijazi, F.; Kosaka, N.; Aboobaker, A.A. Conservation of epigenetic regulation by the MLL3/4 tumour suppressor in planarian pluripotent stem cells. Nat. Commun. 2018, 9, 3633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, K.G.; Molinaro, A.M.; Romero, C.; Dockter, B.; Cable, K.L.; Gonzalez, K.; Zhang, S.; Collins, E.S.; Pearson, B.J.; Zayas, R.M. SoxB1 Activity Regulates Sensory Neuron Regeneration, Maintenance, and Function in Planarians. Dev. Cell. 2018, 47, 331–347.e335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekii, K.; Yorimoto, S.; Okamoto, H.; Nagao, N.; Maezawa, T.; Matsui, Y.; Yamaguchi, K.; Furukawa, R.; Shigenobu, S.; Kobayashi, K. Transcriptomic analysis reveals differences in the regulation of amino acid metabolism in asexual and sexual planarians. Sci. Rep. 2019, 9, 6132. [Google Scholar] [CrossRef] [PubMed]
- Zayas, R.M.; Hernandez, A.; Habermann, B.; Wang, Y.; Stary, J.M.; Newmark, P.A. The planarian Schmidtea mediterranea as a model for epigenetic germ cell specification: Analysis of ESTs from the hermaphroditic strain. Proc. Natl. Acad. Sci. USA 2005, 102, 18491–18496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robb, S.M.; Ross, E.; Sanchez Alvarado, A. SmedGD: The Schmidtea mediterranea genome database. Nucleic Acids Res. 2008, 36, D599–D606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, A.; Li, Y.Q.; Wang, C.; Han, X.S.; Li, G.; Wang, J.Y.; Li, D.S.; Qin, Y.W.; Shi, Y.; Brewer, G.; et al. Heterochromatin protein 1 promotes self-renewal and triggers regenerative proliferation in adult stem cells. J. Cell Biol. 2013, 201, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Almazan, E.M.P.; Lesko, S.L.; Markey, M.P.; Rouhana, L. Girardia dorotocephala transcriptome sequence, assembly, and validation through characterization of piwi homologs and stem cell progeny markers. Dev. Biol. 2018, 433, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Taboada, E.; Moritz, S.; Zeuschner, D.; Stehling, M.; Scholer, H.R.; Salo, E.; Gentile, L. Smed-SmB, a member of the LSm protein superfamily, is essential for chromatoid body organization and planarian stem cell proliferation. Development 2010, 137, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Van Roten, A.; Barakat, A.Z.A.; Wouters, A.; Tran, T.A.; Mouton, S.; Noben, J.P.; Gentile, L.; Smeets, K. A carcinogenic trigger to study the function of tumor suppressor genes in Schmidtea mediterranea. Dis. Model. Mech. 2018, 11, dmm032573. [Google Scholar] [CrossRef] [Green Version]
- Pearson, B.J.; Eisenhoffer, G.T.; Gurley, K.A.; Rink, J.C.; Miller, D.E.; Sanchez Alvarado, A. Formaldehyde-based whole-mount in situ hybridization method for planarians. Dev. Dyn. 2009, 238, 443–450. [Google Scholar] [CrossRef]
- Reddien, P.W.; Sanchez Alvarado, A. Fundamentals of planarian regeneration. Annu. Rev. Cell Dev. Biol. 2004, 20, 725–757. [Google Scholar] [CrossRef] [Green Version]
- Gentile, L.; Cebria, F.; Bartscherer, K. The planarian flatworm: An in vivo model for stem cell biology and nervous system regeneration. Dis. Model. Mech. 2011, 4, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, M.A.; Kim, H.J.; Jejelowo, O.; Pourmand, N. Whole-transcriptome RNAseq analysis from minute amount of total RNA. Nucleic Acids Res. 2011, 39, e120. [Google Scholar] [CrossRef]
- Eisenhoffer, G.T.; Kang, H.; Sanchez Alvarado, A. Molecular analysis of stem cells and their descendants during cell turnover and regeneration in the planarian Schmidtea mediterranea. Cell Stem Cell 2008, 3, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science 2005, 309, 1564–1566. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Wenemoser, D.; Reddien, P.W. Planarian regeneration involves distinct stem cell responses to wounds and tissue absence. Dev. Biol. 2010, 344, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Gurley, K.A.; Rink, J.C.; Sanchez Alvarado, A. Beta-catenin defines head versus tail identity during planarian regeneration and homeostasis. Science 2008, 319, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Agata, K.; Soejima, Y.; Kato, K.; Kobayashi, C.; Umesono, Y.; Watanabe, K. Structure of the planarian central nervous system (CNS) revealed by neuronal cell markers. Zoolog. Sci. 1998, 15, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez Alvarado, A.; Newmark, P.A. Double-stranded RNA specifically disrupts gene expression during planarian regeneration. Proc. Natl. Acad. Sci. USA 1999, 96, 5049–5054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newmark, P.A.; Wang, Y.; Chong, T. Germ cell specification and regeneration in planarians. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.J., 3rd; Hou, X.; Romanova, E.V.; Lambrus, B.G.; Miller, C.M.; Saberi, A.; Sweedler, J.V.; Newmark, P.A. Genome-wide analyses reveal a role for peptide hormones in planarian germline development. PLoS Biol. 2010, 8, e1000509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Stary, J.M.; Wilhelm, J.E.; Newmark, P.A. A functional genomic screen in planarians identifies novel regulators of germ cell development. Genes Dev. 2010, 24, 2081–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurley, K.A.; Elliott, S.A.; Simakov, O.; Schmidt, H.A.; Holstein, T.W.; Sanchez Alvarado, A. Expression of secreted Wnt pathway components reveals unexpected complexity of the planarian amputation response. Dev. Biol. 2010, 347, 24–39. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, C.; Watanabe, K.; Agata, K. The process of pharynx regeneration in planarians. Dev. Biol. 1999, 211, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orii, H.; Ito, H.; Watanabe, K. Anatomy of the planarian Dugesia japonica I. The muscular system revealed by antisera against myosin heavy chains. Zoolog. Sci. 2002, 19, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddien, P.W.; Bermange, A.L.; Murfitt, K.J.; Jennings, J.R.; Sanchez Alvarado, A. Identification of genes needed for regeneration, stem cell function, and tissue homeostasis by systematic gene perturbation in planaria. Dev. Cell 2005, 8, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Molina, M.D.; Salo, E.; Cebria, F. The BMP pathway is essential for re-specification and maintenance of the dorsoventral axis in regenerating and intact planarians. Dev. Biol. 2007, 311, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannini, L.; Rossi, L.; Deri, P.; Gremigni, V.; Salvetti, A.; Salo, E.; Batistoni, R. Djeyes absent (Djeya) controls prototypic planarian eye regeneration by cooperating with the transcription factor Djsix-1. Dev. Biol. 2004, 269, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Cutie, S.; Hoang, A.T.; Payumo, A.Y.; Huang, G.N. Unconventional Functions of Muscles in Planarian Regeneration. Dev. Cell 2017, 43, 657–658. [Google Scholar] [CrossRef] [Green Version]
- Scimone, M.L.; Cote, L.E.; Reddien, P.W. Orthogonal muscle fibres have different instructive roles in planarian regeneration. Nature 2017, 551, 623–628. [Google Scholar] [CrossRef]
- Gross, J.M.; Dowling, J.E. Tbx2b is essential for neuronal differentiation along the dorsal/ventral axis of the zebrafish retina. Proc. Natl. Acad. Sci. USA 2005, 102, 4371–4376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsthoefel, D.J.; James, N.P.; Escobar, D.J.; Stary, J.M.; Vieira, A.P.; Waters, F.A.; Newmark, P.A. An RNAi screen reveals intestinal regulators of branching morphogenesis, differentiation, and stem cell proliferation in planarians. Dev. Cell 2012, 23, 691–704. [Google Scholar] [CrossRef] [Green Version]
- Vij, S.; Rink, J.C.; Ho, H.K.; Babu, D.; Eitel, M.; Narasimhan, V.; Tiku, V.; Westbrook, J.; Schierwater, B.; Roy, S. Evolutionarily ancient association of the FoxJ1 transcription factor with the motile ciliogenic program. PLoS Genet. 2012, 8, e1003019. [Google Scholar] [CrossRef]
- Cowles, M.W.; Brown, D.D.; Nisperos, S.V.; Stanley, B.N.; Pearson, B.J.; Zayas, R.M. Genome-wide analysis of the bHLH gene family in planarians identifies factors required for adult neurogenesis and neuronal regeneration. Development 2013, 140, 4691–4702. [Google Scholar] [CrossRef] [Green Version]
- Iyer, H.; Collins, J.J., 3rd; Newmark, P.A. NF-YB Regulates Spermatogonial Stem Cell Self-Renewal and Proliferation in the Planarian Schmidtea mediterranea. PLoS Genet. 2016, 12, e1006109. [Google Scholar] [CrossRef] [PubMed]
- Owlarn, S.; Bartscherer, K. Go ahead, grow a head! A planarian’s guide to anterior regeneration. Regeneration 2016, 3, 139–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handberg-Thorsager, M.; Salo, E. The planarian nanos-like gene Smednos is expressed in germline and eye precursor cells during development and regeneration. Dev. Genes Evol. 2007, 217, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.A.; Gentile, L. A lineage CLOUD for neoblasts. Semin. Cell Dev. Biol. 2019, 87, 22–29. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Year | Author | Transcriptome Analysis | Species | Sequencing Samples |
|---|---|---|---|---|---|
| 1 | 2010 | Blythe, et al. | 454, SOLiD3 | S. mediterranea | Regenerating fragments at 6, 12, 24, 36, 48, 72, 96, 120, 144 h |
| of anterior and posterior regeneration and intact | |||||
| 2 | 2010 | Abril, et al. | 454 | S. mediterranea | Mixed sample of intact and regenerating planarians (1, 3, 5, and 7 days), |
| Irradiates intact and regenerating animals (1, 3, 5, and 7 days of regeneration) | |||||
| 3 | 2011 | Qin, et al. | Illumina HiseqTM 2000 | D. japonica | Regenerating planarian from Day 1 to Day 10 and intact |
| 4 | 2011 | Adamidi, et al. | 454, Illumina GAIIX | S. mediterranea | Whole animal |
| 5 | 2011 | Sandmann, et al. | Illumina Genome Analyzer II | S. mediterranea | Regenerating head from 0 to 3 days |
| SOLiD3 | S. mediterranea | Regenerating head and tail regions at 0, 1, and 6 h after amputation | |||
| 6 | 2012 | Solana, et al. | SOLiD4 | S. mediterranea | Iraddiate animals at 2, 4, 7 days and wild type intact |
| 7 | 2012 | Shibata, et al. | HiCEP | D. japonica | Intact, Irradiated planarians, Neoblasts from X1/2 fractions |
| 8 | 2012 | Galloni, et al. | DGE | S. mediterranea | Irradiated and normal regenerating samples at 0–7 days after amputation |
| 9 | 2012 | Resch, et al. | Illumina GAIIX | S. mediterranea | non-irradiated or irradiated |
| 10 | 2012 | Nishimura, et al. | Sanger Sequences | D. japonica | Head fragments after amputation |
| 11 | 2012 | Lapan, et al. | Illumina Genome Analyzer II | S. mediterranea | Eyes, amputated heads (above the pharynx and coronal amputation was made to |
| remove dorsal tissues including eye) | |||||
| 12 | 2012 | Labbe, et al. | Illumina HiSeq 2000 | S. mediterranea | FACS sorted stem cells, irradiate animals at 7 days post irradiation |
| 13 | 2012 | Onal, et al. | Illumina Genome Analyzer II | S. mediterranea | X1, X2, Xin |
| 14 | 2013 | Sikes, et al. | Illumina Sequencing | Procotyla fluviatills | Fragments of worms, intact |
| 15 | 2013 | März, et al. | Illumina HiScanSQ | S. mediterranea | Smed-pitx RNAi and control fragments at 3 days post amputation |
| 16 | 2013 | Kao, et al. | SOLiD3 | S. mediterranea | Regenerating head and tail fragments 0, 6, 12, 24, 46, 48, and 72 h after amputation. |
| 454 | G. tigrina | Tail fragments of Smed-Prep RNAi animals 24 h after amputation | |||
| Hofstenia miamia | Embryonic development (2-cell stage through nine days old embryos from multiple parents) | ||||
| 17 | 2014 | Srivastana, et al. | 454 | S. mediterranea | Regeneration at 1, 6, 18, 24, 72 h after amputation |
| 18 | 2014 | Vogg, et al. | Illumina HiScanSQ | S. mediterranea | FoxD RNAi tail stumps at 0 and 3 days post amputation and regenerating control |
| 19 | 2014 | Scimone, et al. | Illumina HiSeq | S. mediterranea | X1 isolation after amputation |
| 20 | 2015 | Reuter, et al. | Illumina HiScanSQ | S. mediterranea | dsRNA notum, 18 h post amputation |
| 21 | 2015 | Wheeler, et al. | Illumina HiSeq 2000 | G. tigrina | Amputation, +serotonin, control |
| 22 | 2016 | Pang, et al. | Illumina HiSeq 2000 | D. japonica | Whole animal |
| 23 | 2016 | Molinaro, et al. | Illumina HiSeq 2500, Single Cell Seq | S. mediterranea | FACS sorted X1, X2 from head |
| 24 | 2018 | Almazan, et al. | Illumina HiSeq 2500 | G. dorotocephala MA-C2 | Intact, 1, 4, days post amputation, Auricles |
| 25 | 2018 | Zeng, et al. | Illumina HiSeq 2500, scRNA-seq | S. mediterranea | Bulk RNA-seq of live cells and fixed cells (X1, X2, Xin), X1 neoblasts (200,000 cells) from |
| wild-type animals, X1 + X2 cells from 7 day sub-lethally irradiated animals | |||||
| 26 | 2018 | Mihaylova, et al. | Illumina NextSeq | S. mediterranea | FACS sorted G2/M cells from knockdown and control RNAi animals 3days of regeneration |
| 27 | 2018 | Ross, et al. | HiSeq 2000 | S. mediterranea | SoxB1-2 RNAi and control RNAi animals at day 6, 14, and 24 after the first RNAI treatment |
| scRNA-seq (Molinaro, et al., 2016) | |||||
| 28 | 2019 | Sekii, et al. | Illumina HiSeq 2000 | D. ryukyuensis | Asexual sample, Sexual sample, Innate Sexual sample |
| 29 | 2020 | Forsthoefel, et al. | Nextseq 500 | S. mediterranea | LCM (medial intestine, lateral intestine, non-intestine) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki-Horiuchi, Y.; Schmitz, H.; Barlassina, C.; Eccles, D.; Sinn, M.; Ortmeier, C.; Moritz, S.; Gentile, L. Transcription Factors Active in the Anterior Blastema of Schmidtea mediterranea. Biomolecules 2021, 11, 1782. https://doi.org/10.3390/biom11121782
Suzuki-Horiuchi Y, Schmitz H, Barlassina C, Eccles D, Sinn M, Ortmeier C, Moritz S, Gentile L. Transcription Factors Active in the Anterior Blastema of Schmidtea mediterranea. Biomolecules. 2021; 11(12):1782. https://doi.org/10.3390/biom11121782
Chicago/Turabian StyleSuzuki-Horiuchi, Yoko, Henning Schmitz, Carlotta Barlassina, David Eccles, Martina Sinn, Claudia Ortmeier, Sören Moritz, and Luca Gentile. 2021. "Transcription Factors Active in the Anterior Blastema of Schmidtea mediterranea" Biomolecules 11, no. 12: 1782. https://doi.org/10.3390/biom11121782
APA StyleSuzuki-Horiuchi, Y., Schmitz, H., Barlassina, C., Eccles, D., Sinn, M., Ortmeier, C., Moritz, S., & Gentile, L. (2021). Transcription Factors Active in the Anterior Blastema of Schmidtea mediterranea. Biomolecules, 11(12), 1782. https://doi.org/10.3390/biom11121782