
Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions

 , , , and
, , , and
Abstract
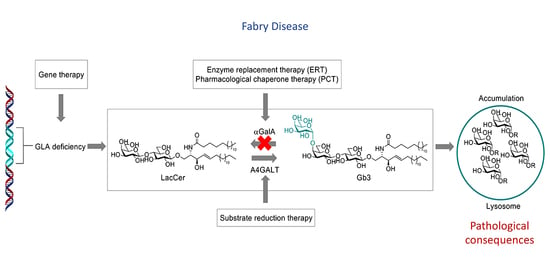
:
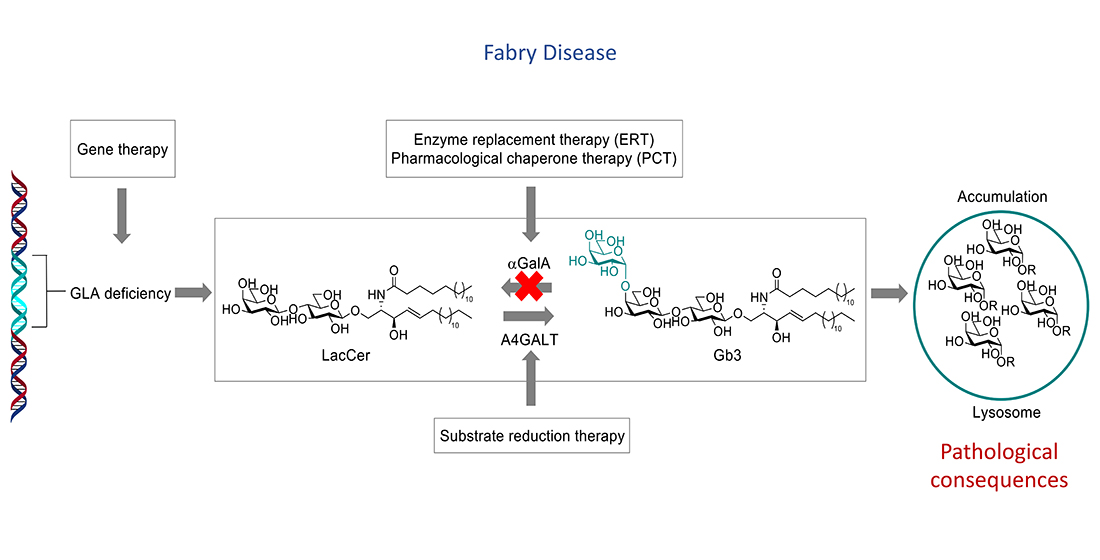{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
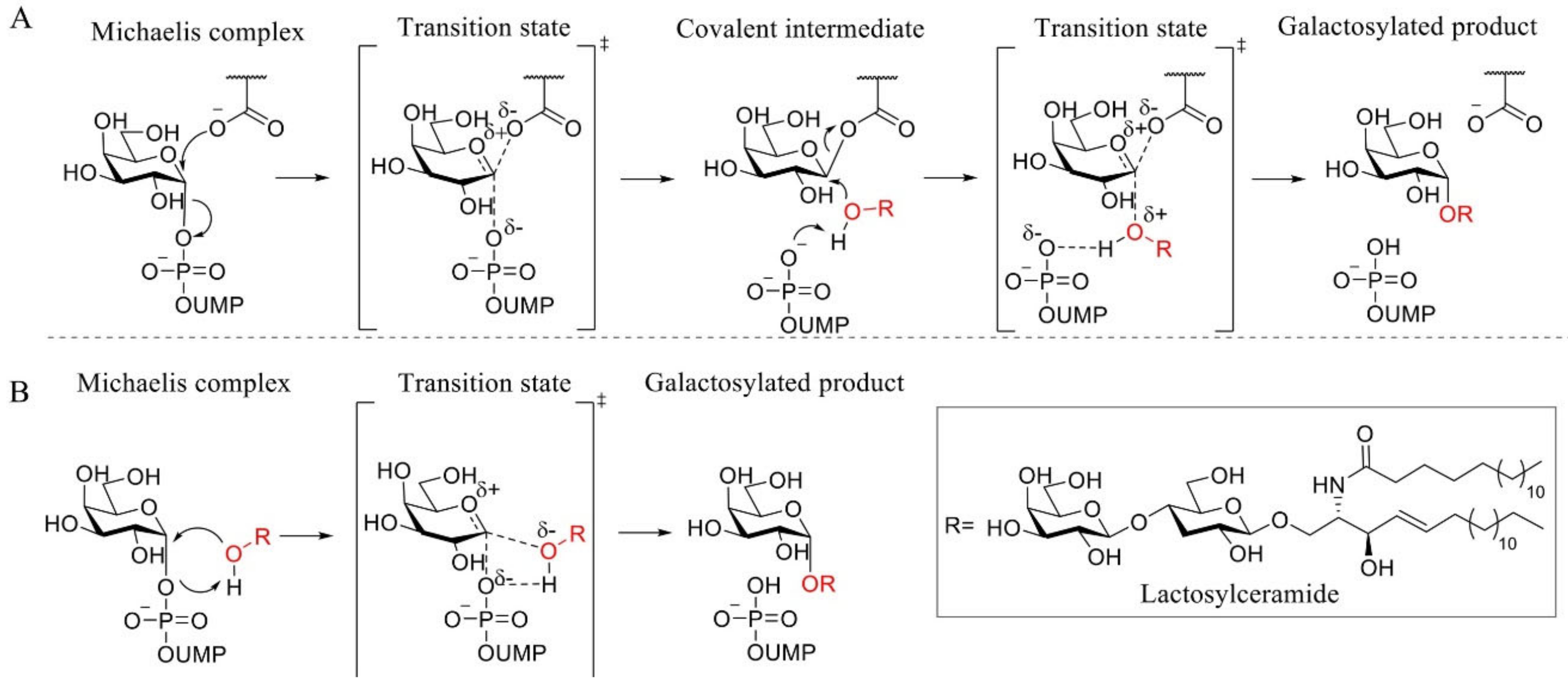{kind=link}
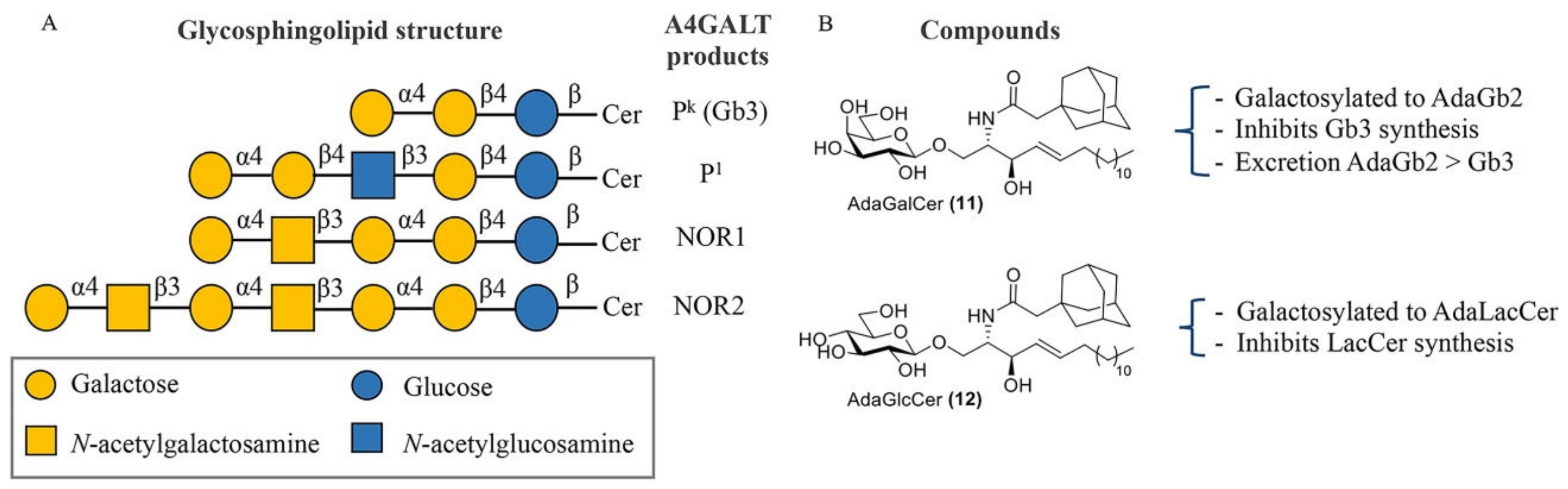{kind=link}
1. Introduction
2. Clinical Manifestation of FD
3. Storage Cells and Secondary Storage Lipids
4. Pathophysiology
5. Diagnosis
6. α-GalA: Reaction Mechanism and Activity-Based Probes (ABPs)
7. Present α-GalA-Centered Therapy Approaches
8. A4GALT: Reaction Mechanism and Enzymatic Products
9. A4GALT Inhibitors and Future Directions
10. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, W. A Case of Angeo-Keratoma. Br. J. Dermatol. 1898, 10, 113–117. [Google Scholar] [CrossRef]
- Fabry, J. Ein Beitrag zur Kenntniss der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). Arch. Dermatol. Res. 1898, 43, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Desnick, R.J.; Ioannou, Y.A. α-Galactosidase a Deficiency. Fabry Disease. The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Sweeley, C.C.; Klionsky, B. Fabry’s Disease: Classification as a sphingolipidosis and partial char-acterization of a novel glycolipid. J. Biol. Chem. 1963, 238, 3148–3150. [Google Scholar] [CrossRef]
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic Defect in Fabry’s Disease. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef]
- Kint, J.A. Fabry’s Disease: Alpha-Galactosidase Deficiency. Science 1970, 167, 1268–1269. [Google Scholar] [CrossRef]
- Hamers, M.N.; Westerveld, A.; Khan, M.; Tager, J.M. Characterization of α-galactosidase isoen-zymes in normal and fabry human-Chinese hamster somatic cell hybrids. Hum. Genet. 1977, 36, 289–297. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.G.; Westerveld, A.; Khan, P.M.; Tager, J.M. Localization of a gene for human α-galactosidase B (=N-Acetyl-α-D-Galactosaminidase) on chromosome 22. Qual. Life Res. 1978, 44, 305–312. [Google Scholar] [CrossRef]
- Sakuraba, H.; Matsuzawa, F.; Aikawa, S.-I.; Doi, H.; Kotani, M.; Nakada, H.; Fukushige, T.; Kanzaki, T. Structural and immunocytochemical studies on α-N-acetylgalactosaminidase deficiency (Schindler/Kanzaki disease). J. Hum. Genet. 2003, 49, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kytidou, K.; Beenakker, T.J.M.; Westerhof, L.B.; Hokke, C.H.; Moolenaar, G.F.; Goosen, N.; Mirzaian, M.; Ferraz, M.J.; de Geus, M.A.R.; Kallemeijn, W.W.; et al. Human Alpha Galactosidases Transiently Produced in Nicotiana benthamiana Leaves: New Insights in Substrate Specificities with Relevance for Fabry Disease. Front. Plant. Sci. 2017, 8, 1026. [Google Scholar] [CrossRef] [Green Version]
- Dean, K.J.; Sweeley, C.C. Studies on human liver α-galactosidases. II. Purification and enzymatic properties of α-galactosidase B (α-N-acetylgalactosaminidase). J. Biol. Chem. 1979, 254, 10001–10005. [Google Scholar] [CrossRef]
- Garman, S.C.; Garboczi, D.N. The Molecular Defect Leading to Fabry Disease: Structure of Human α-Galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef]
- Sakuraba, H. Fabry disease in a Japanese population-molecular and biochemical characteris-tics. Mol. Genet. Metab. Rep. 2018, 17, 73–79. [Google Scholar] [CrossRef]
- Smid, B.E.; Hollak, C.E.M.; Poorthuis, B.J.H.M.; Weerman, M.A.V.D.B.; Florquin, S.; Kok, W.E.M.; Deprez, R.H.L.; Timmermans, J.; Linthorst, G.E. Diagnostic dilemmas in Fabry disease: A case series study on GLA mutations of unknown clinical significance. Clin. Genet. 2015, 88, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M.F.G. Is it Fabry disease? Genet. Med. 2016, 18, 1181–1185. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Natural history of Fabry disease in affected males and obligate carrier females. J. Inherit. Metab. Dis. 2001, 24, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Schachamorov, E.; Beeri, R.; Altarescu, G. X-inactivation in Fabry disease. Gene 2012, 505, 266–268. [Google Scholar] [CrossRef] [PubMed]
- D’Avanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int. J. Mol. Sci. 2020, 21, 1258. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.; Neufeld, E.F. A hypothesis for I-cell disease: Defective hydrolases that do not enter lysosomes. Biochem. Biophys. Res. Commun. 1972, 49, 992–999. [Google Scholar] [CrossRef]
- Schiffmann, R. Fabry disease. Pharmacol. Ther. 2009, 122, 65–77. [Google Scholar] [CrossRef]
- Van der Tol, L.; Smid, B.E.; Poorthuis, B.J.H.M.; Biegstraaten, M.; Deprez, R.H.L.; Linthorst, G.E.; Hollak, C.E.M. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2013, 51, 1–9. [Google Scholar] [CrossRef]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-Y. High incidence of the cardiac variant of Fabry disease revealed by newborn screen-ing in the Taiwan Chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, M.J.; Kallemeijn, W.W.; Mirzaian, M.; Moro, D.H.; Marques, A.R.A.; Wisse, P.; Boot, R.G.; Willems, L.I.; Overkleeft, H.; Aerts, J. Gaucher disease and Fabry disease: New markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Yogasundaram, H.; Kim, D.; Oudit, O.; Thompson, R.B.; Weidemann, F.; Oudit, G.Y. Clinical Features, Diagnosis, and Management of Patients With Anderson-Fabry Cardiomyopathy. Can. J. Cardiol. 2017, 33, 883–897. [Google Scholar] [CrossRef]
- Thurberg, B.L. Globotriaosylceramide accumulation in the Fabry kidney is cleared from mul-tiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedder, A.C.; Strijland, A.; Weerman, M.A.V.B.; Florquin, S.; Aerts, J.M.F.G.; Hollak, C.E.M. Manifestations of Fabry disease in placental tissue. J. Inherit. Metab. Dis. 2006, 29, 106–111. [Google Scholar] [CrossRef]
- Ohshima, T.; Murray, G.J.; Swaim, W.D.; Longenecker, G.; Quirk, J.M.; Cardarelli, C.O.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M.; Brady, R.O.; et al. α-Galactosidase A deficient mice: A model of Fabry disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2540–2544. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.J. α-Galactosidase A-deficient rats accumulate glycosphingolipids and develop car-diorenal phenotypes of Fabry disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 418–429. [Google Scholar]
- Bussink, A.P.; van Eijk, M.; Renkema, G.H.; Aerts, J.M.; Boot, R.G. The Biology of the Gaucher Cell: The Cradle of Human Chitinases. Virus Entry 2006, 252, 71–128. [Google Scholar] [CrossRef]
- Vedder, A.; Cox-Brinkman, J.; Hollak, C.; Linthorst, G.; Groener, J.; Helmond, M.; Scheij, S.; Aerts, J.M. Plasma chitotriosidase in male Fabry patients: A marker for monitoring lipid-laden macrophages and their correction by enzyme replacement therapy. Mol. Genet. Metab. 2006, 89, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Mellett, N.; Hein, L.K.; Brooks, D.A.; Meikle, P.J. Absence of α-galactosidase cross-correction in Fabry heterozygote cultured skin fibroblasts. Mol. Genet. Metab. 2015, 114, 268–273. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Aerts, J.M. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of gly-cosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, M.J.; Marques, A.R.A.; Gaspar, P.; Mirzaian, M.; van Roomen, C.; Ottenhoff, R.; Alfonso, P.; Irún, P.; Giraldo, P.; Wisse, P.; et al. Lyso-glycosphingolipid abnormalities in different murine models of lysosomal storage disorders. Mol. Genet. Metab. 2016, 117, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Togawa, T.; Kodama, T.; Suzuki, T.; Sugawara, K.; Tsukimura, T.; Ohashi, T.; Ishige, N.; Suzuki, K.; Kitagawa, T.; Sakuraba, H. Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Mol. Genet. Metab. 2010, 100, 257–261. [Google Scholar] [CrossRef]
- Krüger, R.; Tholey, A.; Jakoby, T.; Vogelsberger, R.; Mönnikes, R.; Rossmann, H.; Beck, M.; Lackner, K.J. Quantification of the Fabry marker lysoGb3 in human plasma by tandem mass spectrometry. J. Chromatogr. B 2012, 883–884, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Auray-Blais, C. Multiplex tandem mass spectrometry analysis of novel plasma lyso-Gb₃-related analogues in Fabry disease. Anal. Chem. 2014, 86, 3476–3483. [Google Scholar] [CrossRef]
- Talbot, A.; Nicholls, K.; Fletcher, J.M.; Fuller, M. A simple method for quantification of plasma globotriaosylsphingosine: Utility for Fabry disease. Mol. Genet. Metab. 2017, 122, 121–125. [Google Scholar] [CrossRef]
- Rombach, S.; Dekker, N.; Bouwman, M.; Linthorst, G.; Zwinderman, A.; Wijburg, F.; Kuiper, S.; Weerman, M.V.B.; Groener, J.; Poorthuis, B.; et al. Plasma globotriaosylsphingosine: Diagnostic value and relation to clinical manifestations of Fabry disease. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 741–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.J.; Kanack, A.J.; Dahms, N.M. Progress in the understanding and treatment of Fabry disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129437. [Google Scholar] [CrossRef]
- Lücke, T.; Höppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I.P. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M. Altered Sphingolipids Metabolism Damaged Mitochondrial Functions: Lessons Learned From Gaucher and Fabry Diseases. J. Clin. Med. 2020, 9, 1116. [Google Scholar] [CrossRef] [Green Version]
- Ishii, S.; Chang, H.-H.; Kawasaki, K.; Yasuda, K.; Wu, H.-L.; Garman, S.C.; Fan, J.-Q. Mutant α-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F. Fibrosis: A key feature of Fabry disease with potential therapeutic implica-tions. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Shu, L.; Vivekanandan-Giri, A.; Pennathur, S.; Smid, B.E.; Aerts, J.M.; Hollak, C.E.; Shayman, J.A. Establishing 3-nitrotyrosine as a biomarker for the vasculopathy of Fabry disease. Kidney Int. 2014, 86, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Van Eijk, M.; Ferraz, M.J.; Boot, R.G.; Aerts, J.M. Lyso-glycosphingolipids: Presence and consequences. Essays Biochem. 2020, 64, 565–578. [Google Scholar] [CrossRef]
- Choi, L.; Vernon, J.; Kopach, O.; Minett, M.; Mills, K.; Clayton, P.; Meert, T.; Wood, J.N. The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci. Lett. 2015, 594, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Biegstraaten, M.; Hollak, C.E.M.; Bakkers, M.; Faber, C.G.; Aerts, J.M.F.G.; van Schaik, I.N. Small fiber neuropathy in Fabry disease. Mol. Genet. Metab. 2012, 106, 135–141. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D. Globotriaosylsphingosine actions on human glomerular podocytes: Im-plications for Fabry nephropathy. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2011, 26, 1797–1802. [Google Scholar]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaissarian, N.; Kang, J.; Shu, L.; Ferraz, M.J.; Aerts, J.M.; Shayman, J.A. Dissociation of globotriaosylceramide and impaired endothelial function in α-galactosidase-A deficient EA.hy926 cells. Mol. Genet. Metab. 2018, 125, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Rombach, S.M.; Twickler, T.B.; Aerts, J.M.F.G.; Linthorst, G.E.; Wijburg, F.A.; Hollak, C.E. MVasculopathy in patients with Fabry disease: Current controversies and re-search directions. Mol. Genet. Metab. 2010, 99, 99–108. [Google Scholar] [CrossRef]
- Loos, B.; Engelbrecht, A.-M.; Lockshin, R.A.; Klionsky, D.J.; Zakeri, Z. The variability of au-tophagy and cell death susceptibility: Unanswered questions. Autophagy 2013, 9, 1270–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.P. Autophagy-lysosome pathway associated neuropathology and axonal degenera-tion in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol. Commun. 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chévrier, M.; Brakch, N.; Céline, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Laquerrière, A.L.A.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchino, M. A histochemical and electron microscopic study of skeletal and cardiac muscle from a Fabry disease patient and carrier. Acta Neuropathol. 1995, 90, 334–338. [Google Scholar] [CrossRef]
- Aerts, J.M.; Kallemeijn, W.W.; Wegdam, W.; Ferraz, M.J.; van Breemen, M.J.; Dekker, N.; Kramer, G.; Poorthuis, B.J.; Groener, J.E.M.; Cox-Brinkman, J.; et al. Biomarkers in the diagnosis of lysosomal storage disorders: Proteins, lipids, and inhibodies. J. Inherit. Metab. Dis. 2011, 34, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Gold, H.; Mirzaian, M.; Dekker, N.; Ferraz, M.J.; Lugtenburg, J.; Codée, J.D.C.; van der Marel, G.A.; Overkleeft, H.S.; Linthorst, G.E.; Groener, J.E.M.; et al. Quantification of Globotriaosylsphingosine in Plasma and Urine of Fabry Patients by Stable Isotope Ultraperformance Liquid Chromatography–Tandem Mass Spectrometry. Clin. Chem. 2013, 59, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Mirzaian, M. Simultaneous quantitation of sphingoid bases by UPLC-ESI-MS/MS with identi-cal 13 C-encoded internal standards. Clin. Chim. Acta 2017, 466, 178–184. [Google Scholar] [CrossRef]
- Boutin, M.; Lavoie, P.; Abaoui, M.; Auray-Blais, C. Tandem Mass Spectrometry Quantitation of Lyso-Gb 3 and Six Related Analogs in Plasma for Fabry Disease Patients. Curr. Protoc. Hum. Genet. 2016, 90, 17.23.1–17.23.9. [Google Scholar] [CrossRef] [PubMed]
- Polo, G.; Burlina, A.P.; Ranieri, E.; Colucci, F.; Rubert, L.; Pascarella, A.; Burlina, A.B. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphin-golipidoses: A comparative study. Clin. Chem. Lab. Med. 2019, 57, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Houge, G. Fabry or not Fabry-a question of ascertainment. Eur. J. Hum. Genet. EJHG 2011, 19, 1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svarstad, E.; Marti, H.P. The Changing Landscape of Fabry Disease. Clin. J. Am. Soc. Nephrol. 2020, 15, 569–576. [Google Scholar] [CrossRef]
- Bichet, D.G.; Aerts, J.M.; Llm, C.A.-B.; Maruyama, H.; Mehta, A.B.; Skuban, N.; Krusinska, E.; Schiffmann, R. Correction: Assessment of plasma lyso-Gb3 for clinical monitoring of treatment response in migalastat-treated patients with Fabry disease. Genet. Med. 2021, 23, 238. [Google Scholar] [CrossRef]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Tomasic, I.B.; Metcalf, M.C.; Guce, A.I.; Clark, N.E.; Garman, S.C. Interconversion of the spec-ificities of human lysosomal enzymes associated with Fabry and Schindler diseases. J. Biol. Chem. 2010, 285, 21560–21566. [Google Scholar] [CrossRef] [Green Version]
- Crich, D.; Quirke, J.C.K. Glycoside hydrolases restrict the side chain conformation of their sub-strates to gain additional transition state stabilization. J. Am. Chem. Soc. 2020, 142, 16965–16973. [Google Scholar]
- Guce, A.I.; Clark, N.E.; Salgado, E.N.; Ivanen, D.R.; Kulminskaya, A.A.; Brumer, H.; Garman, S.C. Catalytic Mechanism of Human α-Galactosidase. J. Biol. Chem. 2010, 285, 3625–3632. [Google Scholar] [CrossRef] [Green Version]
- Koshland, D.E. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Speciale, G.; Thompson, A.J.; Davies, G.J.; Williams, S.J. Dissecting conformational contribu-tions to glycosidase catalysis and inhibition. Curr. Opin. Struct. Biol. 2014, 28, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Withers, S.G.; Rupitz, K.; Street, I.P. 2-Deoxy-2-fluoro-D-glycosyl fluorides. A new class of specific mechanism-based glycosidase inhibitors. J. Biol. Chem. 1988, 263, 17–20. [Google Scholar] [CrossRef]
- November, F.L. Active site-directed inhibition of galactosidases by conduritol C epoxides (1,2-anhydro-EPI-NEO-inositol). Febs Lett. 1981, 135, 139–144. [Google Scholar]
- Willems, L.I.; Beenakker, T.J.M.; Murray, B.; Gagestein, B.; Elst, H.V.D.; van Rijssel, E.R.; Codee, J.D.C.; Kallemeijn, W.W.; Aerts, J.M.; van der Marel, G.A.; et al. Synthesis of α- and β-Galactopyranose-Configured Isomers of Cyclophellitol and Cyclophellitol Aziridine. Eur. J. Org. Chem. 2014, 2014, 6044–6056. [Google Scholar] [CrossRef]
- Artola, M. α-d-Gal-cyclophellitol cyclosulfamidate is a Michaelis complex analog that stabi-lizes therapeutic lysosomal α-galactosidase A in Fabry disease. Chem. Sci. 2019, 10, 9233–9243. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Armstrong, Z.; Schröder, S.P.; de Boer, C.; Artola, M.; Aerts, J.M.; Overkleeft, H.S.; Davies, G.J. An overview of activity-based probes for glycosidases. Curr. Opin. Chem. Biol. 2019, 53, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Legler, G.; Pohl, S. Synthesis of 5-amino-5-deoxy-d-galactopyranose and 1,5-dideoxy-1,5-imino-d-galactitol, and their inhibition of α- and β-d-galactosidases. Carbohydr. Res. 1986, 155, 119–129. [Google Scholar] [CrossRef]
- Willems, L.I.; Beenakker, T.J.M.; Murray, B.; Scheij, S.; Kallemeijn, W.W.; Boot, R.G.; Verhoek, M.; Donker-Koopman, W.E.; Ferraz, M.J.; van Rijssel, E.R.; et al. Potent and Selective Activity-Based Probes for GH27 Human Retaining α-Galactosidases. J. Am. Chem. Soc. 2014, 136, 11622–11625. [Google Scholar] [CrossRef] [PubMed]
- Kytidou, K.; Beekwilder, J.; Artola, M.; van Meel, E.; Wilbers, R.H.; Moolenaar, G.F.; Aerts, J.M. Nicotiana benthamianaα-galactosidase A1.1 can functionally complement human α-galactosidase A deficiency associated with Fabry disease. J. Biol. Chem. 2018, 293, 10042–10058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogerbrugge, P.; Brouwer, O.; Bordigoni, P.; Cornu, G.; Kapaun, P.; Ortega, J.; O’Meara, A.; Souillet, G.; Frappaz, D.; Blanche, S.; et al. Allogeneic bone marrow transplantation for lysosomal storage diseases. Lancet 1995, 345, 1398–1402. [Google Scholar] [CrossRef]
- Rovelli, A.M. The controversial and changing role of haematopoietic cell transplantation for lyso-somal storage disorders: An update. Bone Marrow Transplant. 2008, 41 (Suppl. 2), S87–S89. [Google Scholar] [CrossRef] [Green Version]
- Brady, R.O. Enzyme replacement therapy: Conception, chaos and culmination. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Aerts, J.M.; Cox, T.M. Roscoe O. Brady: Physician whose pioneering discoveries in lipid bio-chemistry revolutionized treatment and understanding of lysosomal diseases. Blood Cells. Mol. Dis. 2018, 68, 4–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and Efficacy of Recombinant Human α-Galactosidase A Replacement Therapy in Fabry’s Disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffmann, R. Enzyme replacement therapy in fabry disease a randomized controlled trial. J. Am. Med. Assoc. 2001, 285, 2743–2749. [Google Scholar] [CrossRef]
- European Medicines Agency Replagal. European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/product-information/replagal-epar-product-information_en.pdf (accessed on 8 February 2021).
- Blom, D.; Speijer, D.; Linthorst, G.E.; Donker-Koopman, W.G.; Strijland, A.; Aerts, J.M. Recombinant Enzyme Therapy for Fabry Disease: Absence of Editing of Human α-Galactosidase A mRNA. Am. J. Hum. Genet. 2003, 72, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Sakuraba, H. Comparison of the effects of agalsidase alfa and agalsidase beta on cultured hu-man Fabry fibroblasts and Fabry mice. J. Hum. Genet. 2006, 51, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, M.M. Rapid Clathrin-Mediated Uptake of Recombinant α-Gal-A to Lysosome Acti-vates Autophagy. Biomolecules 2020, 10, 837. [Google Scholar] [CrossRef]
- Prabakaran, T.; Nielsen, R.; Larsen, J.V.; Sørensen, S.S.; Rasmussen, U.F.-; Saleem, M.A.; Petersen, C.M.; Verroust, P.J.; Christensen, E.I. Receptor-Mediated Endocytosis of α-Galactosidase A in Human Podocytes in Fabry Disease. PLoS ONE 2011, 6, e25065. [Google Scholar] [CrossRef] [Green Version]
- Priyanka, P.; Parsons, T.B.; Miller, A.; Platt, F.M.; Fairbanks, A.J. Chemoenzymatic Synthesis of a Phosphorylated Glycoprotein. Angew. Chem. Int. Ed. 2016, 55, 5058–5061. [Google Scholar] [CrossRef]
- Tian, W.; Ye, Z.; Wang, S.; Schulz, M.A.; van Coillie, J.; Sun, L.; Chen, Y.-H.; Narimatsu, Y.; Hansen, L.; Kristensen, C.; et al. The glycosylation design space for recombinant lysosomal replacement enzymes produced in CHO cells. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderfer, I.; Shulman, A.; Kizhner, T.; Azulay, Y.; Nataf, Y.; Tekoah, Y.; Shaaltiel, Y. Development and Analytical Characterization of Pegunigalsidase Alfa, a Chemi-cally Cross-Linked Plant Recombinant Human α-Galactosidase-A for Treatment of Fabry Disease. Bioconjug. Chem. 2018, 29, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fab-ry disease, provides sustained plasma concentrations and favorable pharmacodynamics: A 1-year Phase 1/2 clinical trial. J. Inherit. Metab. Dis. 2019, 42, 534–544. [Google Scholar]
- Van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Smid, B.E.; Rombach, S.M.; Aerts, J.M.; Kuiper, S.; Mirzaian, M.; Overkleeft, H.S.; Poorthuis, B.J.H.M.; Hollak, C.E.M.; Groener, J.E.M.; Linthorst, G.E. Consequences of a global enzyme shortage of agalsidase beta in adult Dutch Fabry patients. Orphanet J. Rare Dis. 2011, 6, 69. [Google Scholar] [CrossRef] [Green Version]
- Linthorst, G.E.; Hollak, C.E.M.; Donker-Koopman, W.E.; Strijland, A.; Aerts, J.M.F.G. En-zyme therapy for Fabry disease: Neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004, 66, 1589–1595. [Google Scholar] [CrossRef] [Green Version]
- Sakuraba, H.; Togawa, T.; Tsukimura, T.; Kato, H. Plasma lyso-Gb3: A biomarker for monitoring fabry patients during enzyme replacement therapy. Clin. Exp. Nephrol. 2018, 22, 843–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rombach, S.M.; Aerts, J.M.F.G.; Poorthuis, B.J.H.M.; Groener, J.E.M.; Donker-Koopman, W.; Hendriks, E.; Mirzaian, M.; Kuiper, S.; Wijburg, F.A.; Hollak, C.E.M.; et al. Long-Term Effect of Antibodies against Infused Alpha-Galactosidase A in Fabry Disease on Plasma and Urinary (lyso)Gb3 Reduction and Treatment Outcome. PLoS ONE 2012, 7, e47805. [Google Scholar] [CrossRef] [PubMed]
- Bénichou, B.; Goyal, S.; Sung, C.; Norfleet, A.M.; O’Brien, F. A retrospective analysis of the po-tential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol. Genet. Metab. 2009, 96, 4–12. [Google Scholar] [CrossRef]
- Ishii, S.; Kase, R.; Sakuraba, H.; Suzuki, Y. Characterization of a Mutant α-Galactosidase Gene Product for the Late-Onset Cardiac Form of Fabry Disease. Biochem. Biophys. Res. Commun. 1993, 197, 1585–1589. [Google Scholar] [CrossRef]
- Fan, J.-Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α–galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Ishii, S.; Kizu, H.; Ikeda, K.; Yasuda, K.; Kato, A.; Martin, O.R.; Fan, J.-Q. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. JBIC J. Biol. Inorg. Chem. 2000, 267, 4179–4186. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Migalastat: First Global Approval. Drugs 2016, 76, 1147–1152. [Google Scholar] [CrossRef]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef]
- Benjamin, E.R.; Della-Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, C.; Cardone, M.; Fontana, F.; Rossi, B.; Tuzzi, M.R.; Tarallo, A.; Parenti, G. The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme re-placement therapy in pompe disease fibroblasts. Mol. Ther. 2009, 17, 964–971. [Google Scholar] [CrossRef]
- Pisani, A.; Porto, C.; Andria, G.; Parenti, G. Synergy between the pharmacological chaperone 1-deoxygalactonojirimycin and agalsidase alpha in cultured fibroblasts from patients with Fabry dis-ease. J. Inherit. Metab. Dis. 2014, 37, 145–146. [Google Scholar] [CrossRef]
- Porto, C.; Pisani, A.; Rosa, M.; Acampora, E.; Avolio, V.; Tuzzi, M.R.; Visciano, B.; Gagliardo, C.; Materazzi, S.; la Marca, G.; et al. Synergy between the pharmacological chaperone 1-deoxygalactonojirimycin and the human recombinant alpha-galactosidase A in cultured fibroblasts from patients with Fabry disease. J. Inherit. Metab. Dis. 2011, 35, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.R.; Khanna, R.; Schilling, A.; Flanagan, J.J.; Pellegrino, L.J.; Brignol, N.; Lun, Y.; Guillen, D.; E Ranes, B.; Frascella, M.; et al. Co-administration With the Pharmacological Chaperone AT1001 Increases Recombinant Human α-Galactosidase A Tissue Uptake and Improves Substrate Reduction in Fabry Mice. Mol. Ther. 2012, 20, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnock, D.G.; Bichet, D.G.; Holida, M.; Goker-Alpan, O.; Nicholls, K.; Thomas, M.; Eyskens, F.; Shankar, S.; Adera, M.; Sitaraman, S.; et al. Oral Migalastat HCl Leads to Greater Systemic Exposure and Tissue Levels of Active α-Galactosidase A in Fabry Patients when Co-Administered with Infused Agalsidase. PLoS ONE 2015, 10, e0134341. [Google Scholar] [CrossRef]
- Citro, V.; Peña-García, J.; Den-Haan, H.; Pérez-Sánchez, H.; del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an Allosteric Binding Site on Human Lysosomal Alpha-Galactosidase Opens the Way to New Pharmacological Chaperones for Fabry Disease. PLoS ONE 2016, 11, e0165463. [Google Scholar] [CrossRef] [Green Version]
- Smid, B.E.; Ferraz, M.J.; Verhoek, M.; Mirzaian, M.; Wisse, P.; Overkleeft, H.S.; Hollak, C.E.; Aerts, J.M. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J. Rare Dis. 2016, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterschmitt, M.J.; Crawford, N.P.S.; Gaemers, S.J.M.; Ji, A.J.; Sharma, J.; Pham, T.T. Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Oral Venglustat in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2021, 10, 86–98. [Google Scholar] [CrossRef]
- Guérard, N.; Morand, O.; Dingemanse, J. Lucerastat, an iminosugar with potential as substrate reduction therapy for glycolipid storage disorders: Safety, tolerability, and pharmacokinetics in healthy subjects. Orphanet J. Rare Dis. 2017, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, M.; Huston, M.W.; Pagant, S.; Gan, L.; St. Martin, S.; Sproul, S.; Richards, D.; Ballaron, S.; Hettini, K.; Ledeboer, A.; et al. AAV2/6 Gene Therapy in a Murine Model of Fabry Disease Results in Supraphysiological Enzyme Activity and Effective Substrate Reduction. Mol. Ther. Methods Clin. Dev. 2020, 18, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Huston, M.W.; Yasuda, M.; Pagant, S.; St Martin, S.; Cao, L.; Falese, L.; Wechsler, T. Liver-targeted AAV gene therapy vectors produced by a clinical scale manu-facturing process result in high, continuous therapeutic levels of enzyme activity and effective sub-strate reduction in mouse model of Fabry disease. Mol. Genet. Metab. 2019, 126, S77. [Google Scholar] [CrossRef]
- Kia, A.; McIntosh, J.; Rosales, C.; Hosseini, P.; Sheridan, R.; Spiewak, J.; Mills, K.; Corbau, R.; Nathwani, A.C. Efficacy Evaluation of Liver-Directed Gene Therapy in Fabry Mice. Blood 2018, 132, 2209. [Google Scholar] [CrossRef]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Sims, K.; Politei, J.; Banikazemi, M.; Lee, P. Stroke in Fabry disease frequently occurs before di-agnosis and in the absence of other clinical events: Natural history data from the Fabry Registry. Stroke 2009, 40, 788–794. [Google Scholar] [CrossRef] [Green Version]
- Lairson, L.L.; Chiu, C.P.; Ly, H.D.; He, S.; Wakarchuk, W.W.; Strynadka, N.C.; Withers, S.G. Intermediate Trapping on a Mutant Retaining α-Galactosyltransferase Identifies an Unexpected Aspartate Residue. J. Biol. Chem. 2004, 279, 28339–28344. [Google Scholar] [CrossRef] [Green Version]
- Persson, K. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria men-ingitidis in complex with donor and acceptor sugar analogs. Nat. Struct. Biol. 2001, 8, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Ardèvol, A.; Rovira, C. Reaction Mechanisms in Carbohydrate-Active Enzymes: Glycoside Hy-drolases and Glycosyltransferases. Insights from ab Initio Quantum Mechanics/Molecular Mechan-ics Dynamic Simulations. J. Am. Chem. Soc. 2015, 137, 7528–7547. [Google Scholar] [CrossRef] [PubMed]
- Ardèvol, A.; Rovira, C. The Molecular Mechanism of Enzymatic Glycosyl Transfer with Retention of Configuration: Evidence for a Short-Lived Oxocarbenium-Like Species. Angew. Chem. Int. Ed. 2011, 50, 10897–10901. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Pastores, G.M. Eliglustat for Gaucher’s disease: Trippingly on the tongue. Lancet 2015, 385, 2328–2330. [Google Scholar] [CrossRef]
- Lukas, J.; Giese, A.-K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional Characterisation of Alpha-Galactosidase A Mutations as a Basis for a New Classification System in Fabry Disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wennekes, T.; Berg, R.J.B.H.N.V.D.; Boot, R.G.; van der Marel, G.A.; Overkleeft, H.S.; Aerts, J.M. Glycosphingolipids-Nature, Function, and Pharmacological Modulation. Angew. Chem. Int. Ed. 2009, 48, 8848–8869. [Google Scholar] [CrossRef]
- Steffensen, R.; Carlier, K.; Wiels, J.; Levery, S.B.; Stroud, M.; Cedergren, B.; Clausen, H. Cloning and Expression of the Histo-blood Group Pk UDP-galactose:Galbeta 1-4Glcbeta 1-Cer alpha 1,4-Galactosyltransferase. J. Biol. Chem. 2000, 275, 16723–16729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, Y.; Fukumoto, S.; Furukawa, K.; Okajima, T.; Wiels, J.; Yokoyama, K.; Suzuki, Y.; Urano, T.; Ohta, M.; Furukawa, K. Molecular Cloning of Globotriaosylceramide/CD77 Synthase, a Glycosyltransferase That Initiates the Synthesis of Globo Series Glycosphingolipids. J. Biol. Chem. 2000, 275, 15152–15156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, R.; Duk, M.; Szymczak, K.; Korchagina, E.; Tyborowska, J.; Mikolajczyk, K.; Bovin, N.; Szewczyk, B.; Jaskiewicz, E.; Czerwinski, M. Human Gb3/CD77 synthase reveals specificity toward two or four different acceptors depending on amino acid at position 211, creating Pk, P1 and NOR blood group antigens. Biochem. Biophys. Res. Commun. 2016, 470, 168–174. [Google Scholar] [CrossRef]
- Kaczmarek, R.; Mikolajewicz, K.; Szymczak, K.; Duk, M.; Majorczyk, E.; Krop-Watorek, A.; Buczkowska, A.; Czerwinski, M. Evaluation of an amino acid residue critical for the specificity and activity of human Gb3/CD77 synthase. Glycoconj. J. 2016, 33, 963–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellberg, Å.; Schmidt-Melbye, A.-C.; Reid, M.E.; Olsson, M.L. Expression of a novel missense mutation found in the A4GALT gene of Amish individuals with the p phenotype. Transfusion 2008, 48, 479–487. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Chang, C.-F.; Lin, H.-C.; Lin, K.-S.; Lin, K.-T.; Hung, C.-M.; Lin, T.-M. Functional characterisation of a complex mutation in the α(1,4)galactosyltransferase gene in Taiwanese individuals with p phenotype. Transfus. Med. 2010, 21, 84–89. [Google Scholar] [CrossRef]
- Reymond, D.; Karmali, M.A.; Clarke, I.; Winkler, M.; Petric, M. Comparison of the Western blot assay with the neutralizing-antibody and enzyme-linked immunosorbent assays for measuring anti-body to Verocytotoxin 1. J. Clin. Microbiol. 1997, 35, 609–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proulx, F.; Seidman, E.G.; Karpman, D. Pathogenesis of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Pediatr. Res. 2001, 50, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of Shiga tox-in and ricin. Physiol. Rev. 1996, 346, 99–102. [Google Scholar]
- Cilmi, S.A.; Karalius, B.J.; Choy, W.; Smith, R.N.; Butterton, J.R. Fabry Disease in Mice Protects against Lethal Disease Caused by Shiga Toxin–Expressing EnterohemorrhagicEscherichia coli. J. Infect. Dis. 2006, 194, 1135–1140. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Muneeruddin, K.; Choi, M.Y.; Tao, L.; Bhuiyan, R.H.; Ohmi, Y.; Furukawa, K.; Furukawa, K.; Boland, S.; Shaffer, S.A.; et al. Genome-wide CRISPR screens for Shiga toxins and ricin reveal Golgi proteins critical for glycosylation. PLoS Biol. 2018, 16, e2006951. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, T.; Sekizuka, T.; Tachida, Y.; Sakuma, C.; Morimoto, K.; Kuroda, M.; Hanada, K. A CRISPR Screen Identifies LAPTM4A and TM9SF Proteins as Glycolipid-Regulating Factors. iScience 2019, 11, 409–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloster, T.M.; Vocadlo, D.J. Developing inhibitors of glycan processing enzymes as tools for enabling glycobiology. Nat. Chem. Biol. 2012, 8, 683–694. [Google Scholar] [CrossRef]
- Kamani, M.; Mylvaganam, M.; Tian, R.; Rigat, B.; Binnington, B.; Lingwood, C. Adamantyl Glycosphingolipids Provide a New Approach to the Selective Regulation of Cellular Glycosphingolipid Metabolism. J. Biol. Chem. 2011, 286, 21413–21426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantom, P.A.; Coward, J.K.; Blanchard, J.S. UDP-(5F)-GlcNAc acts as a slow-binding inhibitor of MshA, a retaining glycosyltransferase. J. Am. Chem. Soc. 2010, 132, 6626–6627. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.C.T.; Jiang, S.; Rush, J.S.; Waechter, A.C.J.; Coward, J.K. Glycosyltransferase Mechanisms: Impact of a 5-Fluoro Substituent in Acceptor and Donor Substrates on Catalysis. Biochemistry 2007, 46, 11630–11638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaluddin, H.; Tumbale, P.; Withers, S.G.; Acharya, K.R.; Brew, K. Conformational Changes Induced by Binding UDP-2F-galactose to α-1,3 Galactosyltransferase- Implications for Catalysis. J. Mol. Biol. 2007, 369, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.-C.; Kwon, Y.-G.; Kim, D.-H.; Jang, I.-S.; Cho, J.-W.; Chung, S.-K. Chemoenzymatic syntheses of carbasugar analogues of nucleoside diphosphate sugars: UDP-carba-Gal, UDP-carba-GlcNAc, UDP-carba-Glc, and GDP-carba-Man. Chem. Commun. 2009, 1733–1735, 1733–1735. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.L.; Tian, F.; Lee, L.V.; Wong, C.H. Synthesis and evaluation of transition-state ana-logue inhibitors of α-1,3-fucosyltransferase. Angew. Chem. Int. Ed. 2002, 114, 3167–3170. [Google Scholar] [CrossRef]
- Descroix, K.; Pesnot, T.; Yoshimura, Y.; Gehrke, S.S.; Wakarchuk, W.W.; Palcic, M.M.; Wagner, G.K. Inhibition of Galactosyltransferases by a Novel Class of Donor Analogues. J. Med. Chem. 2012, 55, 2015–2024. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Frische, K. A new galactosyl transferase inhibitor. Bioorganic Med. Chem. Lett. 1993, 3, 1747–1750. [Google Scholar] [CrossRef]
- Wagstaff, B.A.; Rejzek, M.; Pesnot, T.; Tedaldi, L.M.; Caputi, L.; O’Neill, E.C.; Field, R. A Enzymatic synthesis of nucleobase-modified UDP-sugars: Scope and limita-tions. Carbohydr. Res. 2015, 404, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesnot, T.; Jørgensen, R.; Palcic, M.M.; Wagner, G.K. Structural and mechanistic basis for a new mode of glycosyltransferase inhibition. Nat. Chem. Biol. 2010, 6, 321–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kok, K.; Zwiers, K.C.; Boot, R.G.; Overkleeft, H.S.; Aerts, J.M.F.G.; Artola, M. Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules 2021, 11, 271. https://doi.org/10.3390/biom11020271
Kok K, Zwiers KC, Boot RG, Overkleeft HS, Aerts JMFG, Artola M. Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules. 2021; 11(2):271. https://doi.org/10.3390/biom11020271
Chicago/Turabian StyleKok, Ken, Kimberley C. Zwiers, Rolf G. Boot, Hermen S. Overkleeft, Johannes M. F. G. Aerts, and Marta Artola. 2021. "Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions" Biomolecules 11, no. 2: 271. https://doi.org/10.3390/biom11020271
APA StyleKok, K., Zwiers, K. C., Boot, R. G., Overkleeft, H. S., Aerts, J. M. F. G., & Artola, M. (2021). Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules, 11(2), 271. https://doi.org/10.3390/biom11020271