
Involvement of Cellular Prion Protein in Invasion and Metastasis of Lung Cancer by Inducing Treg Cell Development


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cancer Development
2.3. Scrapie Infection
2.4. Quantitative PCR Analysis
2.5. Confocal Images
2.6. Flow Cytometry
2.7. Statistical Analysis
3. Results
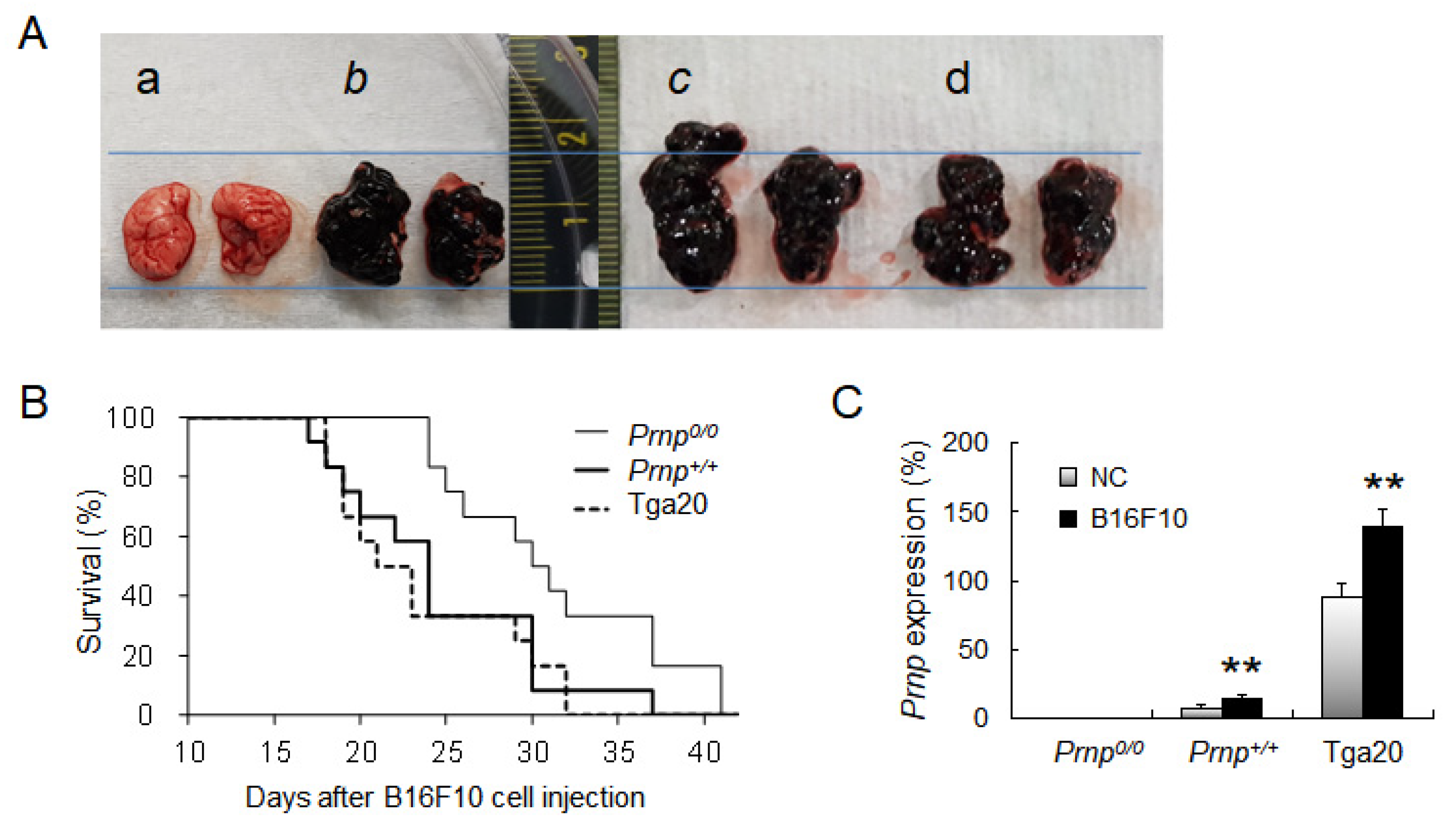
3.1. Effect of PrPC Expression on Lung Cancer Development
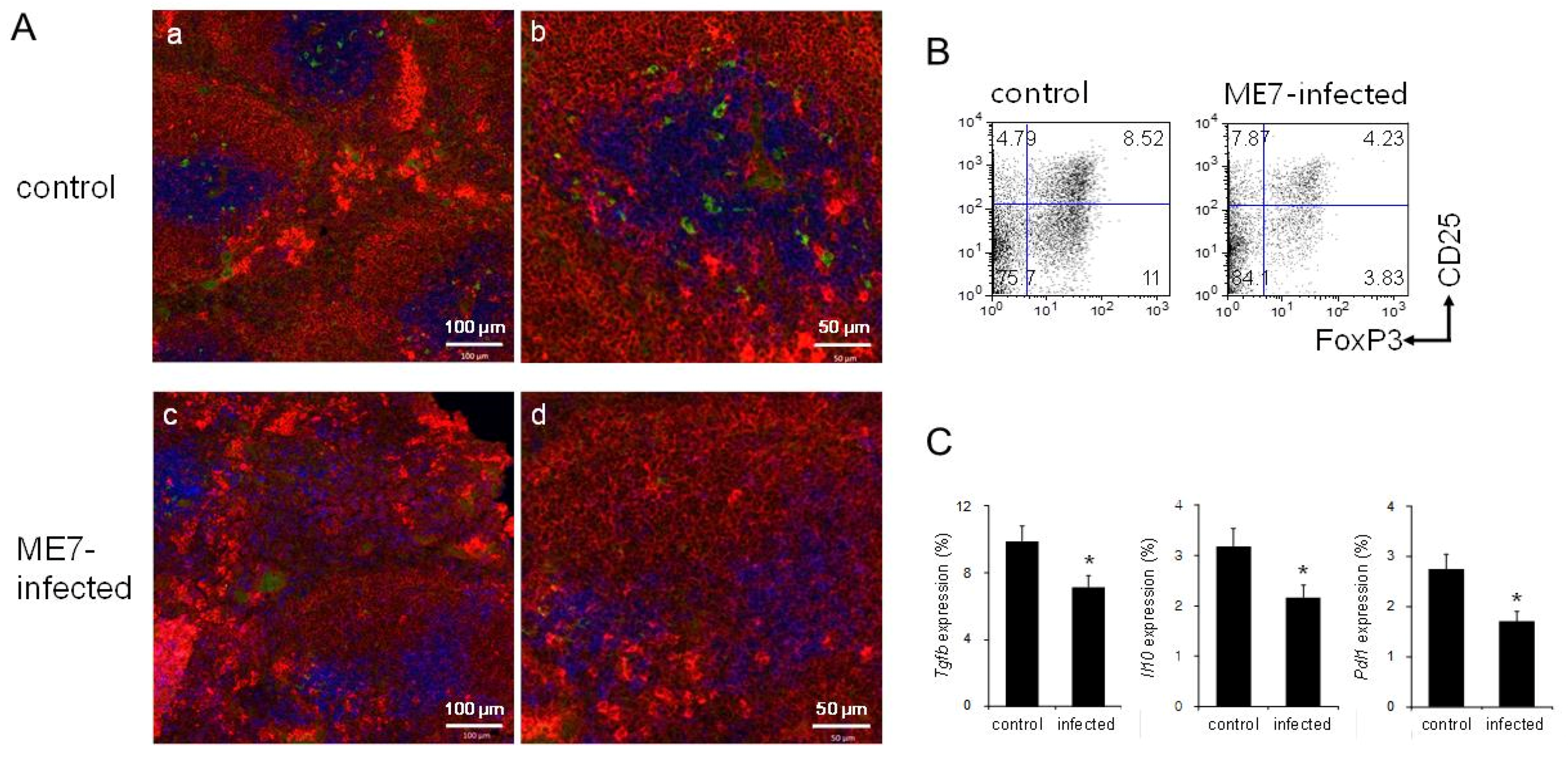
3.2. Increased Numbers of Treg Cells in Prnp+/+ and Tga20 Mice
3.3. Increased Expression of TGF-β and PD-L1 in Tga20 Mice
3.4. No Effect of PrPC Expression on Natural Killer or CD8 T Cell Numbers
3.5. Decreased Numbers of Treg Cells by introducing PrPSc through ME7 Scrapie Infection
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PrPC | cellular prion protein |
| GPI | glycosylphosphatidylinositol |
| PrPSc | pathogenic prion protein |
| Prnp0/0 | PrP-deficient |
| Tga20 | PrP-overexpressing |
| TGF-β | transforming growth factor-beta |
| IL-10 | interleukin-10 |
| Treg | regulatory T |
| FoxP3 | forkhead box P3 |
| PD-L1 | programmed death ligand-1 |
| PD1 | programmed cell death protein 1 |
| ERK | extracellular signal-regulated kinase |
| MMP-9 | metalloprotease-9 |
| NF-κB | nuclear factor-κB |
| actb | beta-actin |
| IgM | immunoglobulin M |
| NK | natural killer |
| NC | negative control |
| B16F10 | B16F10 melanoma cells |
| DMEM | Dulbecco’s modified Eagle medium |
| PBS | phosphate-buffered saline |
| FITC | fluorescein isothiocyanate |
| APC | allophycocyanin |
References
- Mabbott, N.A.; Brown, K.L.; Manson, J.; Bruce, M.E. T-lymphocyte activation and the cellular form of the prion protein. Immunology 1997, 92, 161–165. [Google Scholar] [CrossRef]
- Antoine, N.; Cesbron, J.Y.; Coumans, B.; Jolois, O.; Zorzi, W.; Heinen, E. Differential expression of cellular prion protein on human blood and tonsil lymphocytes. Haematologica 2000, 85, 475–480. [Google Scholar]
- Cashman, N.R.; Loertscher, R.; Nalbantoglu, J.; Shaw, I.; Kascsak, R.J.; Bolton, D.C.; Bendheim, P.E. Cellular isoform of the scrapie agent protein participates in lymphocyte activation. Cell 1990, 61, 185–192. [Google Scholar] [CrossRef]
- Kim, S.; Han, S.; Lee, Y.E.; Jung, W.-J.; Lee, H.S.; Kim, Y.-S.; Choi, E.-K.; Kim, M.-Y. Prion protein-deficient mice exhibit decreased CD4 T and LTi cell numbers and impaired spleen structure. Immunobiology 2016, 221, 94–102. [Google Scholar] [CrossRef]
- Kim, S.; Han, S.; Lee, H.S.; Kim, Y.S.; Choi, E.K.; Kim, M.Y. Impaired spleen structure and chemokine expression in ME7 scrapie-infected mice. Immunobiology 2016, 221, 871–878. [Google Scholar] [CrossRef]
- Kim, S.; Han, S.; Kim, T.; Nam, J.; Kim, Y.-S.; Choi, E.-K.; Kim, M.-Y. Prolonged follicular helper T cell responses in ME7 scrapie-infected mice. Prion 2018, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Lakkaraju, A.K.; Frontzek, K. Toward Therapy of Human Prion Diseases. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 331–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Makarava, N.; Katorcha, E.; Savtchenko, R.; Brossmer, R.; Baskakov, I.V. Post-conversion sialylation of prions in lymphoid tissues. Proc. Natl. Acad. Sci. USA 2015, 112, E6654–E6662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roucou, X.; LeBlanc, A.C. Cellular prion protein neuroprotective function: Implications in prion diseases. J. Mol. Med. 2005, 83, 3–11. [Google Scholar] [PubMed]
- Camandola, S.; Cutler, R.G.; Gary, D.S.; Milhavet, O.; Mattson, M.P. Suppression of calcium release from inositol 1,4,5-trisphosphate-sensitive stores mediates the anti-apoptotic function of nuclear factor-kappaB. J. Biol. Chem. 2005, 280, 22287–22296. [Google Scholar] [CrossRef] [Green Version]
- Mattei, V.; Garofalo, T.; Misasi, R.; Circella, A.; Manganelli, V.; Lucania, G.; Pavan, A.; Sorice, M. Prion protein is a component of the multimolecular signaling complex involved in T cell activation. FEBS Lett. 2004, 560, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Stella, R.; Massimino, M.L.; Sorgato, M.C.; Bertoli, A. Prion and TNFalpha: TAC(E)it agreement between the prion protein and cell signaling. Cell Cycle 2010, 9, 4616–4621. [Google Scholar] [CrossRef] [Green Version]
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Peng, M.; Chen, L.; Yang, X.; Li, H.; Shi, R.; Wu, G.; Cai, L.; Song, Q.; Li, C. Prion Protein Protects Cancer Cells against Endoplasmic Reticulum Stress Induced Apoptosis. Virol. Sin. 2019, 34, 222–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roucou, X.; Gains, M. Neuroprotective functions of prion protein. J. Neurosci. Res. 2003, 75, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-M.; Choi, E.-K.; Carp, R.I.; Kim, Y.-S. Oxidative stress impairs autophagic flux in prion protein-deficient hippocampal cells. Autophagy 2012, 8, 1448–1461. [Google Scholar] [CrossRef] [Green Version]
- Timmons, J.J.; Cohessy, S.; Wong, E.T. Injection of Syngeneic Murine Melanoma Cells to Determine Their Metastatic Potential in the Lungs. J. Vis. Exp. 2016, e54039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, J.; Wu, G.; Zhang, J.; Li, H.; Gao, S.; Shao, M.; Gao, Z.; Sy, M.-S.; Cao, Y.; Yang, X.; et al. Melanoma migration is promoted by prion protein via Akt-hsp27 signaling axis. Biochem. Biophys. Res. Commun. 2020, 523, 375–381. [Google Scholar] [CrossRef]
- Sollazzo, V.; Galasso, M.; Volinia, S.; Carinci, F. Prion proteins (PRNP and PRND) are over-expressed in osteosarcoma. J. Orthop. Res. 2011, 30, 1004–1012. [Google Scholar] [CrossRef]
- Gil, M.; Kim, Y.K.; Kim, K.-E.; Kim, W.; Park, C.-S.; Lee, K.J. Cellular prion protein regulates invasion and migration of breast cancer cells through MMP-9 activity. Biochem. Biophys. Res. Commun. 2016, 470, 213–219. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, S.; Huang, D.; Cui, M.; Hu, H.; Zhang, L.; Wang, W.; Parameswaran, N.; Jackson, M.; Osborne, B.; et al. Cellular Prion Protein Mediates Pancreatic Cancer Cell Survival and Invasion through Association with and Enhanced Signaling of Notch1. Am. J. Pathol. 2016, 186, 2945–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Pan, Y.; Ning, X.; Sun, L.; Lan, M.; Hong, L.; Du, J.; Liu, N.; Liu, C.; Qiao, T.; et al. Overexpression of PrPC and Its Antiapoptosis Function in Gastric Cancer. Tumor Biol. 2006, 27, 84–91. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pot, C.; Apetoh, L.; Kuchroo, V.K. Type 1 regulatory T cells (Tr1) in autoimmunity. Semin. Immunol. 2011, 23, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Bi, Y.; Sun, K.; Wang, C. IL-10-producing type 1 regulatory T cells and allergy. Cell. Mol. Immunol. 2007, 4, 269–275. [Google Scholar]
- Colombo, M.P.; Piconese, S. Regulatory T-cell inhibition versus depletion: The right choice in cancer immunotherapy. Nat. Rev. Cancer 2007, 7, 880–887. [Google Scholar] [CrossRef]
- A Ward-Hartstonge, K.; A Kemp, R. Regulatory T-cell heterogeneity and the cancer immune response. Clin. Transl. Immunol. 2017, 6, e154. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Regulatory T cell subsets in human cancer: Are they regulating for or against tumor progression? Cancer Immunol. Immunother. 2014, 63, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, S.F. FOXP3: Of Mice and Men. Annu. Rev. Immunol. 2006, 24, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Christoffersson, G.; von Herrath, M. Regulatory Immune Mechanisms beyond Regulatory T Cells. Trends Immunol. 2019, 40, 482–491. [Google Scholar] [CrossRef]
- Thompson, C.; Powrie, F. Regulatory T cells. Curr. Opin. Pharmacol. 2004, 4, 408–414. [Google Scholar] [CrossRef]
- Byrne, W.L.; Mills, K.H.; Lederer, J.A.; O’Sullivan, G.C. Targeting Regulatory T Cells in Cancer. Cancer Res. 2011, 71, 6915–6920. [Google Scholar] [CrossRef] [Green Version]
- Golgher, D.; Jones, E.; Powrie, F.; Elliott, T.; Gallimore, A. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur. J. Immunol. 2002, 32, 3267–3275. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Dahm-Vicker, M.; Simon, A.K.; Green, A.; Powrie, F.; Cerundolo, V.; Gallimore, A. Depletion of CD25+ regulatory cells results in suppression of melanoma growth and induction of autoreactivity in mice. Cancer Immun. 2002, 2, 1. [Google Scholar]
- Yu, P.; Lee, Y.; Liu, W.; Krausz, T.; Chong, A.; Schreiber, H.; Fu, Y.X. Intratumor depletion of CD4+ cells unmasks tumor im-munogenicity leading to the rejection of late-stage tumors. J. Exp. Med. 2005, 201, 779–791. [Google Scholar]
- Büeler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.-P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nat. Cell Biol. 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Rülicke, T.; Raeber, A.; Sailer, A.; Moser, M.; Oesch, B.; Brandner, S.; Aguzzi, A.; Weissmann, C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996, 15, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Gaspal, F.M.; Wiggett, H.E.; McConnell, F.M.; Gulbranson-Judge, A.; Raykundalia, C.; Walker, L.S.; Goodall, M.D.; Lane, P.J. CD4(+)CD3(-) accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where T cells collab-orate with B cells. Immunity 2003, 18, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Han, S.; Kim, M.Y. Effects of interleukin-15 on human CD3(-)CD117(+)CD56(-)OX40L(+) cell differentiation. Hum. Immunol. 2010, 71, 745–750. [Google Scholar] [CrossRef]
- Chen, X.; Du, Y.; Lin, X.; Qian, Y.; Zhou, T.; Huang, Z. CD4 + CD25 + regulatory T cells in tumor immunity. Int. Immunopharmacol. 2016, 34, 244–249. [Google Scholar] [CrossRef]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Huntington, N.D.; Smyth, C.G.M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Bearss, D.J.; Browne, L.W.; Calaluce, R.; Nagle, R.B.; Von Hoff, D.D. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res. 2002, 62, 2890–2896. [Google Scholar] [PubMed]
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Giambelluca, M.A.; Morelli, L.; Frati, A.; Fornai, F. The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer. Cancers 2021, 13, 170. [Google Scholar] [CrossRef]
- Wiegmans, A.P.; Saunus, J.M.; Ham, S.; Lobb, R.J.; Kutasovic, J.R.; Dalley, A.J.; Miranda, M.; Atkinson, C.; Foliaki, S.T.; Ferguson, K.; et al. Secreted cellular prion protein binds doxorubicin and correlates with anthracycline resistance in breast cancer. JCI Insight 2019, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yu, S.; Nakamura, F.; Yin, S.; Xu, J.; Petrolla, A.A.; Singh, N.; Tartakoff, A.; Abbott, D.W.; Xin, W.; et al. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J. Clin. Investig. 2009, 119, 2725–2736. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Pentikainen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.S. Pro-prion binds filamin A, facilitating its interaction with integrin beta1, and contributes to melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhu, T.; Zhao, R. Filamin A regulates EGFR/ERK/Akt signaling and affects colorectal cancer cell growth and migration. Mol. Med. Rep. 2019, 20, 3671–3678. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, M.; Bai, G.; Li, X.; Sun, Z.; Yang, J.; Wang, L.; Sun, J. Filamin A inhibits tumor progression through regulating BRCA1 expression in human breast cancer. Oncol. Lett. 2018, 16, 6261–6266. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene su-perfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derer, A.; Spiljar, M.; Bäumler, M.; Hecht, M.; Fietkau, R.; Frey, B.; Gaipl, U.S. Chemoradiation Increases PD-L1 Expression in Certain Melanoma and Glioblastoma Cells. Front. Immunol. 2016, 7, 610. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Actb | CGTGAAAAGATGACCCAGATCA | TGGTACGACCAGAGGCATACAG |
| Prnp | ATGGCGAACCTTGGCTACTG | CCTGAGGTGGGTAACGGTTG |
| Tgfb | CCGCAACAACGCCATCTATG | CCCGAATGTCTGACGTATTGAAG |
| Pdl1 | GCTCCA AAGGACTTGTACGTG | TGATCTGAAGGGCAGCATTTC |
| Il10 | ACAGCCGGGAAGACAATAACT | GCAGCTCTAGGAGCATGTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cha, S.; Sin, M.-J.; Kim, M.-J.; Kim, H.-J.; Kim, Y.-S.; Choi, E.-K.; Kim, M.-Y. Involvement of Cellular Prion Protein in Invasion and Metastasis of Lung Cancer by Inducing Treg Cell Development. Biomolecules 2021, 11, 285. https://doi.org/10.3390/biom11020285
Cha S, Sin M-J, Kim M-J, Kim H-J, Kim Y-S, Choi E-K, Kim M-Y. Involvement of Cellular Prion Protein in Invasion and Metastasis of Lung Cancer by Inducing Treg Cell Development. Biomolecules. 2021; 11(2):285. https://doi.org/10.3390/biom11020285
Chicago/Turabian StyleCha, Seunghwa, Mi-Ji Sin, Mo-Jong Kim, Hee-Jun Kim, Yong-Sun Kim, Eun-Kyoung Choi, and Mi-Yeon Kim. 2021. "Involvement of Cellular Prion Protein in Invasion and Metastasis of Lung Cancer by Inducing Treg Cell Development" Biomolecules 11, no. 2: 285. https://doi.org/10.3390/biom11020285
APA StyleCha, S., Sin, M. -J., Kim, M. -J., Kim, H. -J., Kim, Y. -S., Choi, E. -K., & Kim, M. -Y. (2021). Involvement of Cellular Prion Protein in Invasion and Metastasis of Lung Cancer by Inducing Treg Cell Development. Biomolecules, 11(2), 285. https://doi.org/10.3390/biom11020285