
p62/SQSTM1/Keap1/NRF2 Axis Reduces Cancer Cells Death-Sensitivity in Response to Zn(II)–Curcumin Complex

 ,
,  and
and 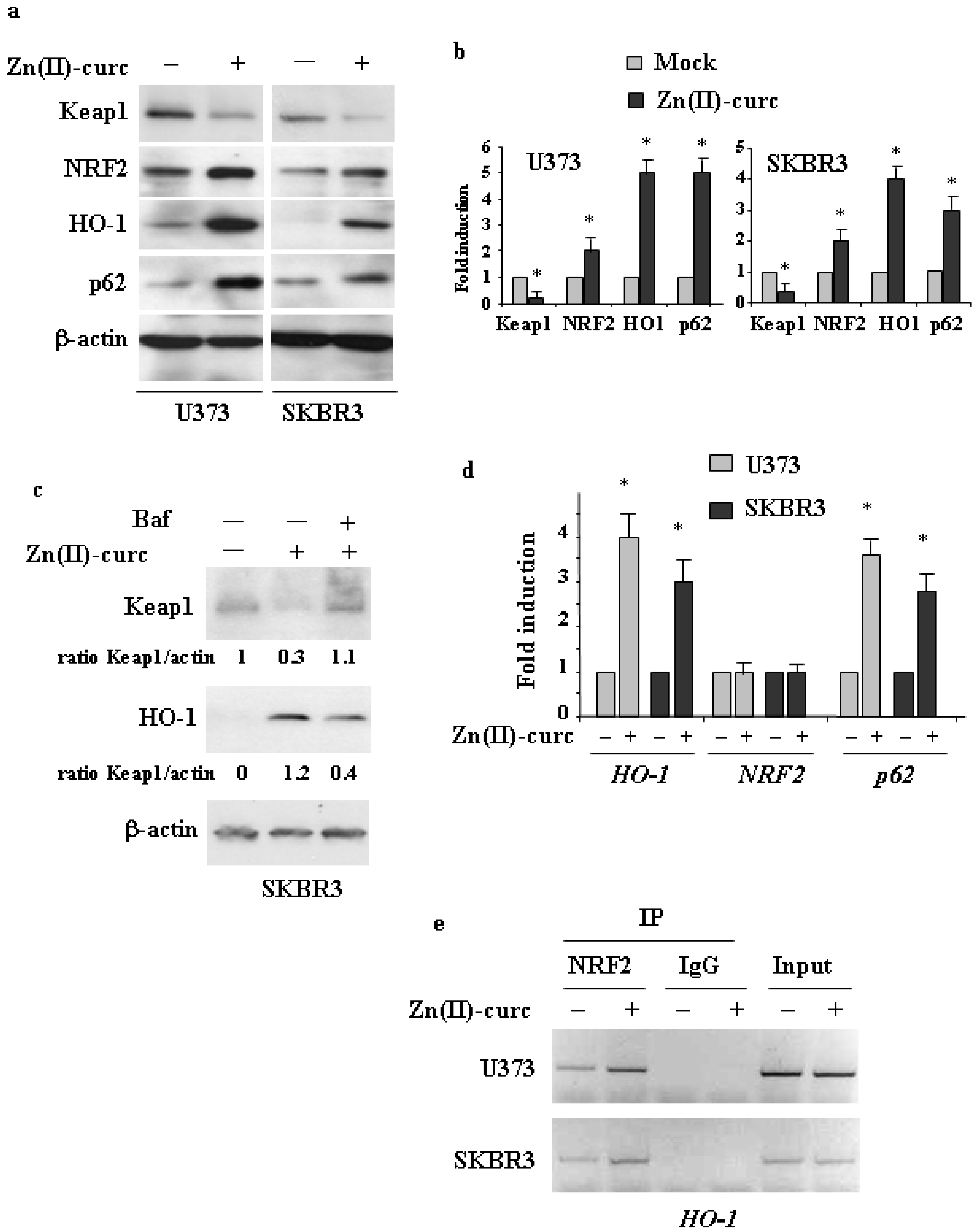{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibodies and Reagents
2.3. Western Blotting
2.4. siRNA Interference
2.5. Cell Viability Assays
2.6. Chromatin Immunoprecipitation (ChIP) Assay
2.7. RNA Extraction and Semiquantitative Reverse Transcription (RT)-PCR Analysis
2.8. Statistical Analysis
3. Results and Discussion
3.1. Induction of the NRF2 Pathway by Zn(II)–Curc
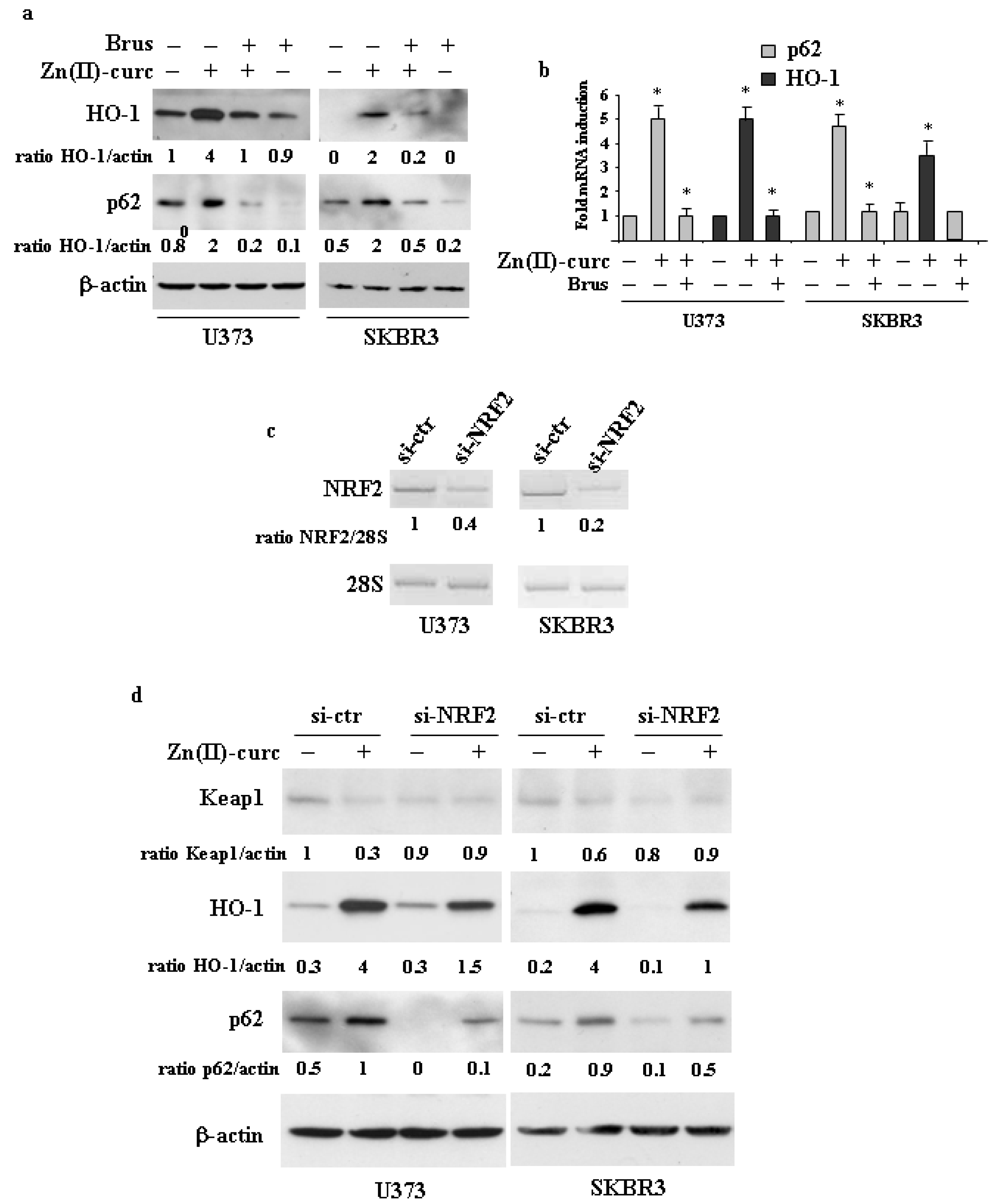
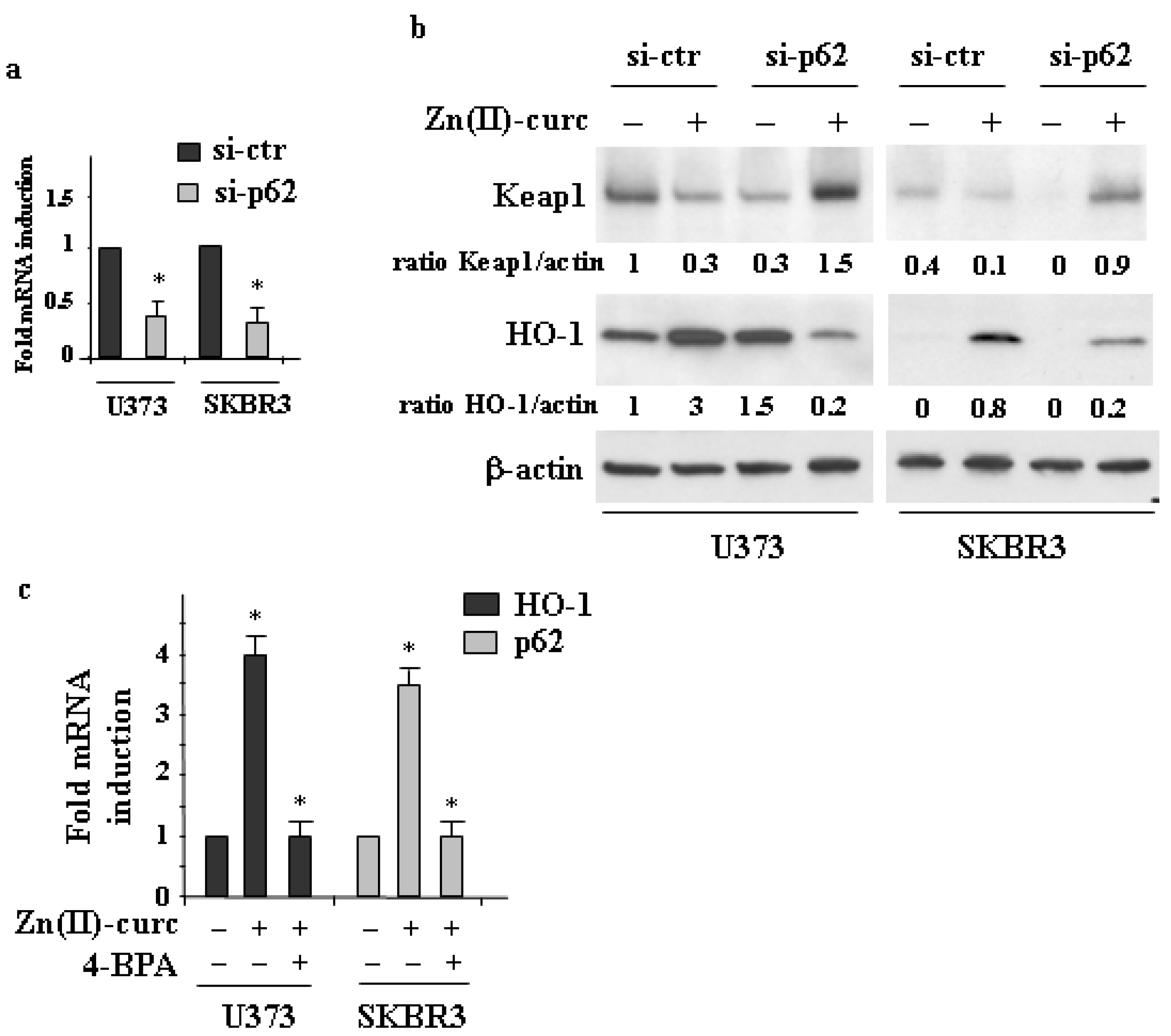
3.2. The Interplay between NRF2 and p62 in Cancer Cells Treated by Zn(II)–Curc
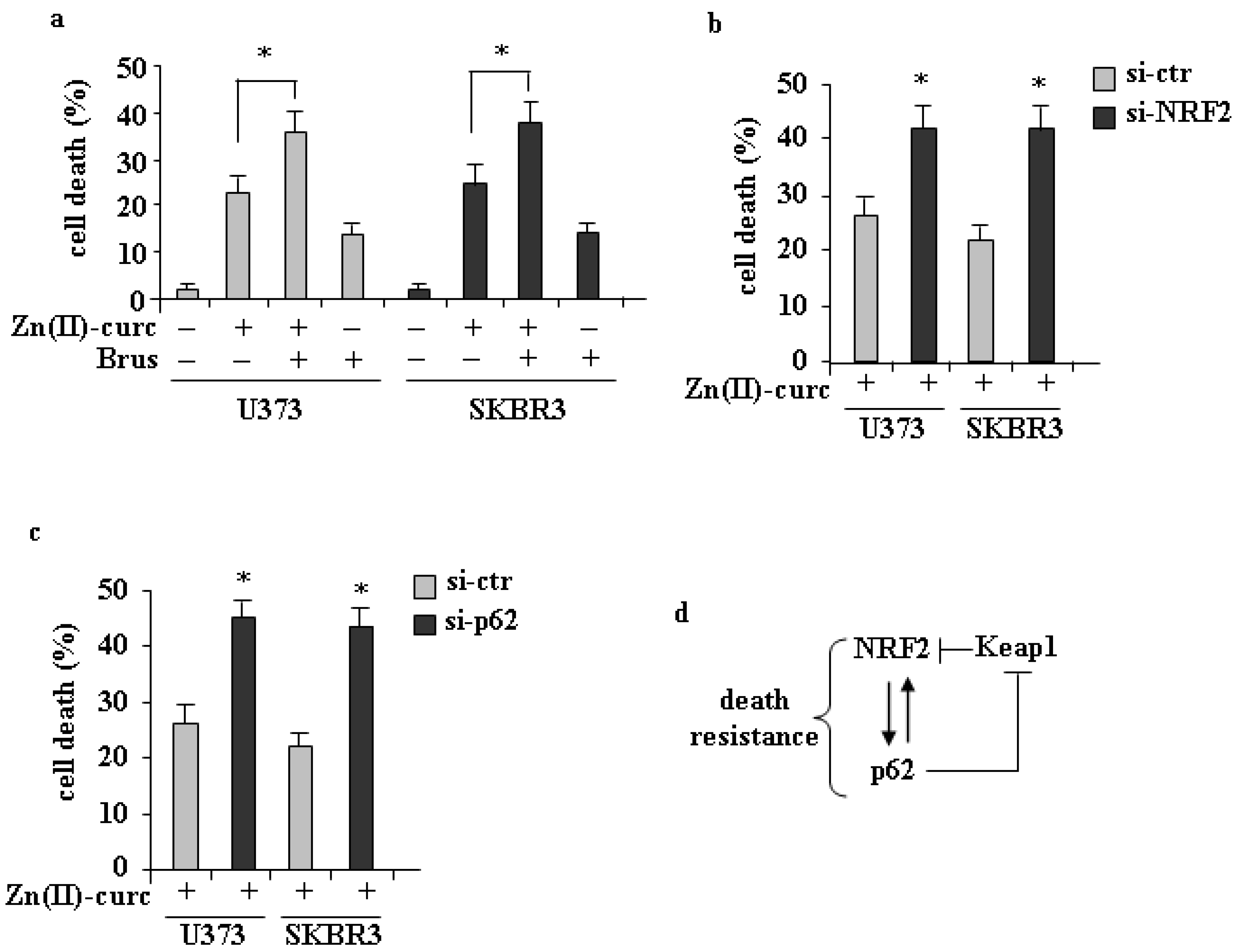
3.3. Role of p62/NRF2 Axis in Zn(II)–Curc-Induced Cell Death
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and hallmarks of cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- No, J.H.; Kim, Y.B.; Song, Y.S. Targeting Nrf2 signaling to combat chemoresistance. J. Cancer Prev. 2014, 19, 111–117. [Google Scholar] [CrossRef] [PubMed]
- McMahonm, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhand, D.D. p62 links autophagy and Nrf2 signaling. Free Rad. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Dayalan Naidu, S.; Kostov, R.V.; Dinkova-Kostova, A.T. Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol. Sci. 2015, 36, 6–14. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of NRF2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. Pharmacological applications of Nrf2 inhibitors as potential antineoplastic drugs. Int. J. Mol. Sci. 2019, 20, 2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; Traversi, G.; Gilardini Montani, M.S.; D’Orazi, V.; Pistritto, G.; Cirone, M.; D’Orazi, G. Reduced chemotherapeutic sensitivity in high glucose condition: Implication of antioxidant response. Oncotarget 2019, 10, 4691–4702. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Baldari, S.; Pettinari, R.; GilardiniMontani, M.S.; D’Orazi, V.; Pistritto, G.; Crispini, A.; Giorno, E.; Toietta, G.; Marchetti, F.; et al. A ruthenium(II) curcumin compound modulates NRF2 expression balancing the cell death/survival outcome in both wild-type and mutant p53-carrying cancer cells. J. Exp. Clin. Cancer Res. 2020, 39, 122. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in ageing and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumor suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Trisciuoglio, D.; Porru, M.; Leonetti, C.; Stoppacciaro, A.; D’Orazi, V.; Avantaggiati, M.L.; Crispini, A.; Pucci, D.; D’Orazi, G. A fluorescent curcumin-based Zn(II)-complex reactivates mutant (R175H and R273H) p53 in cancer cells. J. Exp. Clin. Cancer Res. 2013, 32, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; D’Orazi, V.; Pucci, D.; Cirone, M.; Avantaggiati, M.L.; D’Orazi, G. Mutant p53H175 protein is targeted by Zn(II) compound for degradation through autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; D’Orazi, V.; Crispini, A.; D’Orazi, G. Zn(II)-curc targets p53 in thyroid cancer cells. Int. J. Oncol. 2015, 47, 1241–1248. [Google Scholar] [CrossRef] [Green Version]
- Loh, S.N. The missing Zinc: p53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; Porru, M.; Simon, A.J.; Rechavi, G.; Leonetti, C.; Givol, D.; D’Orazi, G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to ancticancer drugs. Cell Cycle 2011, 10, 1679–1689. [Google Scholar] [CrossRef]
- D’Orazi, G.; Givol, D. p53 reactivation: The link to zinc. Cell Cycle 2012, 11, 2581–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margalit, O.; Simon, A.J.; Yakubov, E.; Puca, R.; Yosepovich, A.; Avivi, C.; Jacob-Hirsch, J.; Gelernter, I.; Harmelin, A.; Barshack, I.; et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int. J. Cancer. 2012, 131, E562–E568. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Garufi, A.; Di Renzo, L.; Granato, M.; Faggioni, A.; D’Orazi, G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunology 2013, 2, e26198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; Ubertini, V.; Mancini, F.; D’Orazi, V.; Baldari, S.; Moretti, F.; Bossi, G.; D’Orazi, G. The beneficial effect of Zinc(II) on low-dose chemotherapeutic sensitivity involves p53 activation in wild-type p53 cancer cells. J. Exp. Clin. Cancer Res. 2015, 34, 87. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; Trisciuoglio, D.; Cirone, M.; D’Orazi, G. ZnCl2 sustains the adriamycin-induced cell death inhibited by high glucose. Cell Death Dis. 2016, 7, e2280. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.R.; Wu, Y.S.; Tunki, L.; Chellian, R.; Halmuthur, M.S.; Muller, S.; Pandy, V. What Has Come out from Phytomedicines and Herbal Edibles for the Treatment of Cancer? Chem. Med. Chem. 2018, 13, 1854–1872. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Ahmadi, Z.; Mohammadinejad, R.; Farkhondeh, T.; Samarghandian, S. Curcumin activates the Nrf2 pathway and induces cellular protection against oxidative injury. Curr. Mol. Med. 2020, 20, 116–133. [Google Scholar] [CrossRef]
- D’Orazi, G.; Garufi, A.; Cirone, M. NRF2 interferes with HIPK2/p53 activity to impair solid tumors chemosensitivity. IUBMB Life 2020, 72, 1634–1639. [Google Scholar]
- Pucci, D.; Crispini, A.; Mendiguchía, B.S.; Pirillo, S.; Ghedini, M.; Morelli, S.; Bartolo, L.D. Improving the bioactivity of Zn(II)-curcumin based complexes. Dalton Trans. 2013, 42, 9679–9687. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granato, M.; Santarelli, R.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Garufi, A.; Trivedi, P.; Frati, L.; D’Orazi, G.; Faggioni, A.; et al. JNK and macroautophagy activation by bortezomib has a pro-survival effect of in primary effusion lymphoma cells. PLoS ONE 2013, 8, e75965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; Federici, G.; Gilardini Montani, M.S.; Crispini, A.; Cirone, M.; D’Orazi, G. Interplay between endoplasmic reticulum (ER) stress and autophagy by Zn(II)-curc induces mutant p53H273 degradation. Biomolecules 2020, 10, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilardini Montani, M.S.; Cecere, N.; Granato, M.; Romeo, M.A.; Falcinelli, L.; Ciciarelli, U.; D’Orazi, G.; Faggioni, A.; Cirone, M. Mutant p53, stabilized by its interplay with HSP90, activates a positive feed-back loop between NRF2 and p62 that induces chemo-resistance to Apigenin in pancreatic cancer cells. Cancers 2019, 11, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Orazi, G.; Cirone, M. Mutant p53 and cellular stress pathways: A criminal alliance that promotes cancer progression. Cancers 2019, 11, 614. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1/Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Char, J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Sanchez-Martin, P.; Saito, T.; Komatsu, M. p62/SQSTM1: “Jack of all trades” in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Hayden, A.; Douglas, J.; Sommerlad, M.; Andrews, L.; Gould, K.; Hussain, S.; Thomas, G.J.; Packham, G.; Crabb, S.J. The NRF2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol. Oncol. 2014, 32, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Onodera, Y.; Motohashi, H.; Takagi, K.; Miki, Y.; Shibahara, Y.; Watanabe, M.; Ishida, T.; Hirakawa, H.; Sasano, H.; Yamamoto, M.; et al. NRF2 immunolocalization in human breast cancer patients as a prognostic factor. Endocr. Relat. Cancer 2014, 21, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Xu, H.; Zhang, B.; Hong, B.; Yan, W.; Zhang, J. Impact of nuclear factor erythroid-derived 2–like 2 and p62/sequestosome expression on prognosis of patients with gliomas. Hum. Pathol. 2015, 46, 843–849. [Google Scholar] [CrossRef]
- Ma, J.Q.; Tuersun, H.; Jiao, S.J.; Zheng, J.H.; Xiao, J.B.; Hasim, A. Functional Role of NRF2 in Cervical Carcinogenesis. PLoS ONE 2015, 10, e0133876. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Ishigami, S.; Arigami, T.; Uenosono, Y.; Yanagita, S.; Uchikado, Y.; Kita, Y.; Nishizono, Y.; Okumura, H.; Nakajo, A.; et al. Clinicopathological significance of nuclear factor (erythroid-2)-related factor 2 (NRF2) expression in gastric cancer. BMC Cancer 2015, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, H.J.; Bao, Q.C.; Wang, L.; Guo, T.K.; Chen, W.L.; Xu, L.L.; Zhou, H.S.; Bian, J.L.; Yang, Y.R.; et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016, 7, 73593–73606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Komatsu, M. Activation of p62/SQTSTM1-Keap1-Nuclear Factor Erythroid 2-Related Factor 2 pathway in cancer. Front. Oncol. 2018, 8, 210. [Google Scholar] [CrossRef]
- Tsai, T.F.; Cen, P.C.; Lin, Y.C.; Chou, K.Y.; Chen, H.E.; Ho, C.Y.; Lin, J.F.; Hwang, T.I. Miconazole Contributes to NRF2 Activation by Noncanonical p62-KEAP1 Pathway in Bladder Cancer Cells. Drug Des. Dev. Ther. 2020, 14, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- Torrente, L.; Maan, G.; Rezig, A.O.; Quinn, J.; Jackson, A.; Grilli, A.; Casares, L.; Zhang, Y.; Kulesskiy, E.; Saarela, J.; et al. High NRF2 levels correlates with poor prognosis in colorectal cancer patients and with sensitivity to the kinase inhibitor AT9283 in vitro. Biomolecules 2020, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ma, D.; Wang, P.; Pan, C.; Fang, Q.; Wang, J. Nrf2 overexpression increases risk of high tumor mutation burden in acute myeloid leukaemia by inhibiting MSH2. Cell Death Dis. 2021, 12, 20. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garufi, A.; Giorno, E.; Gilardini Montani, M.S.; Pistritto, G.; Crispini, A.; Cirone, M.; D’Orazi, G. p62/SQSTM1/Keap1/NRF2 Axis Reduces Cancer Cells Death-Sensitivity in Response to Zn(II)–Curcumin Complex. Biomolecules 2021, 11, 348. https://doi.org/10.3390/biom11030348
Garufi A, Giorno E, Gilardini Montani MS, Pistritto G, Crispini A, Cirone M, D’Orazi G. p62/SQSTM1/Keap1/NRF2 Axis Reduces Cancer Cells Death-Sensitivity in Response to Zn(II)–Curcumin Complex. Biomolecules. 2021; 11(3):348. https://doi.org/10.3390/biom11030348
Chicago/Turabian StyleGarufi, Alessia, Eugenia Giorno, Maria Saveria Gilardini Montani, Giuseppa Pistritto, Alessandra Crispini, Mara Cirone, and Gabriella D’Orazi. 2021. "p62/SQSTM1/Keap1/NRF2 Axis Reduces Cancer Cells Death-Sensitivity in Response to Zn(II)–Curcumin Complex" Biomolecules 11, no. 3: 348. https://doi.org/10.3390/biom11030348
APA StyleGarufi, A., Giorno, E., Gilardini Montani, M. S., Pistritto, G., Crispini, A., Cirone, M., & D’Orazi, G. (2021). p62/SQSTM1/Keap1/NRF2 Axis Reduces Cancer Cells Death-Sensitivity in Response to Zn(II)–Curcumin Complex. Biomolecules, 11(3), 348. https://doi.org/10.3390/biom11030348