
TRIM32: A Multifunctional Protein Involved in Muscle Homeostasis, Glucose Metabolism, and Tumorigenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
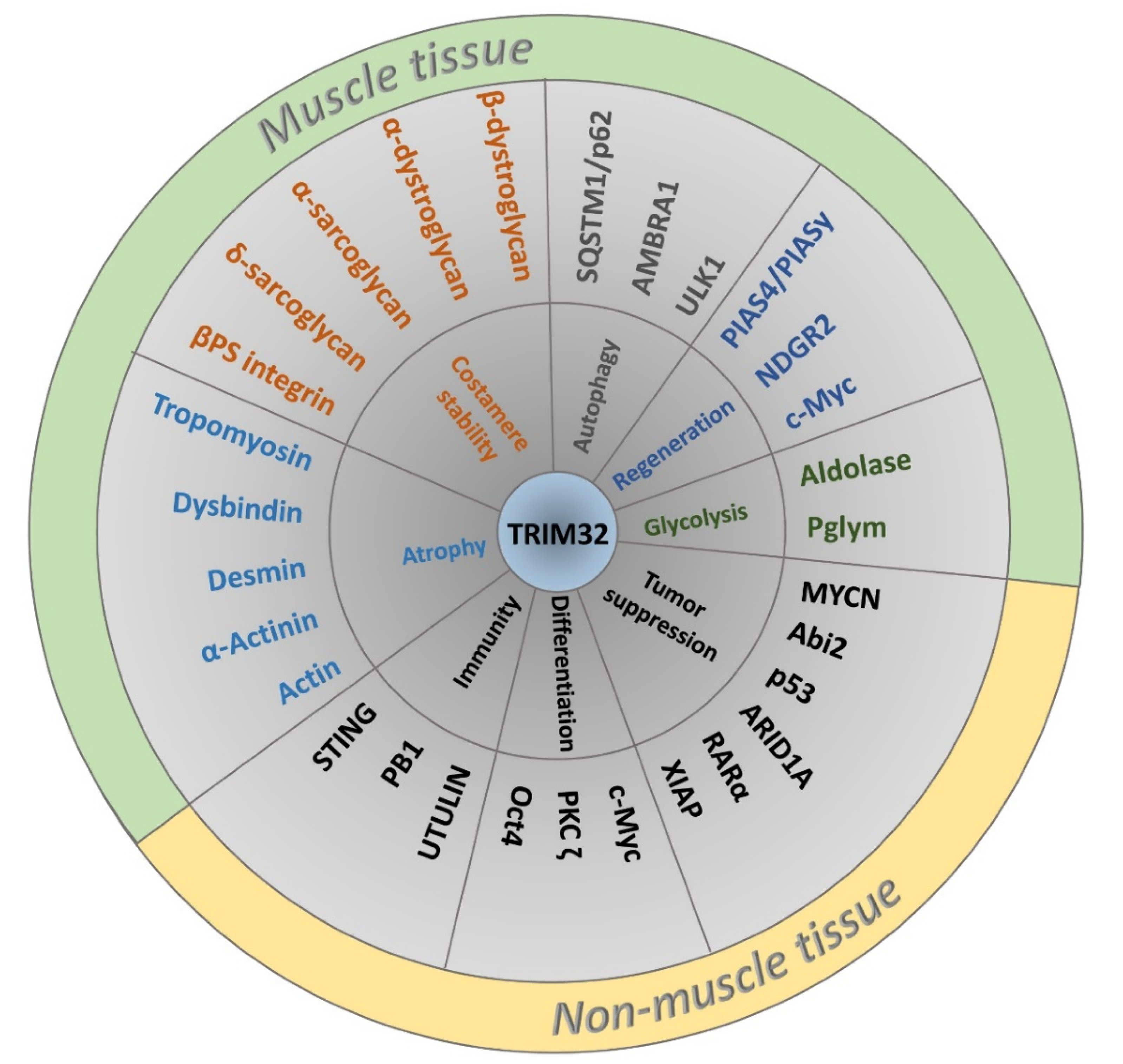
Abstract
:1. The TRIM Family of Proteins
1.1. Structural and Functional Organization of TRIM32
1.2. E3 Ligase Activity and TRIM32 Substrates
2. Pathomechanism of TRIM32 in Limb-Girdle Muscular Dystrophy Type 2H
2.1. Mutations Associated with TRIM32
2.2. In Vitro Assessment of TRIM32 Function
2.3. Modeling of LGMD2H Point Mutations
2.4. TRIM32 Mouse Models for LGMD2H
2.5. Drosophila Model for LGMD2H
3. The Complicated Role of TRIM32 in Muscle Homeostasis
3.1. Atrophy
3.2. Regeneration and Growth
4. TRIM32, Glucose Metabolism, and Link to Cancer
4.1. Glycolytic Enzymes in Muscle Physiology
4.2. Drosophila as a Model for Glycolytic Growth
4.3. TRIM32 Function as a Tumor Suppressor
4.4. TRIM32 and Its Oncogenic Role in Tumor Growth
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef] [Green Version]
- Tocchini, C.; Ciosk, R. TRIM-NHL proteins in development and disease. Semin. Cell Dev. Biol. 2015, 47-48, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Wulczyn, F.G.; Cuevas, E.; Franzoni, E.; Rybak, A. miRNAs Need a Trim: Regulation of miRNA Activity by Trim-NHL Proteins. Adv. Exp. Med. Biol. 2011, 700, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 2017, 161, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.P.; Haubrich, K.; Perez-Borrajero, C.; Hennig, J. Emerging RNA-binding roles in the TRIM family of ubiquitin ligases. Biol. Chem. 2019, 400, 1443–1464. [Google Scholar] [CrossRef] [PubMed]
- Fridell, R.A.; Harding, L.S.; Bogerd, H.P.; Cullen, B.R. Identification of a novel human zinc finger protein that specifically interacts with the activation domain of lentiviral Tat proteins. Virology 1995, 209, 347–357. [Google Scholar] [CrossRef]
- Lazzari, E.; Meroni, G. TRIM32 ubiquitin E3 ligase, one enzyme for several pathologies: From muscular dystrophy to tumours. Int. J. Biochem. Cell Biol. 2016, 79, 469–477. [Google Scholar] [CrossRef]
- Shieh, P.B.; Kudryashova, E.; Spencer, M.J. Limb-girdle muscular dystrophy 2H and the role of TRIM32. Handb. Clin. Neurol. 2011, 101, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Frosk, P.; Weiler, T.; Nylen, E.; Sudha, T.; Greenberg, C.R.; Morgan, K.; Fujiwara, T.M.; Wrogemann, K. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am. J. Hum. Genet. 2002, 70, 663–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domsch, K.; Ezzeddine, N.; Nguyen, H.T. Abba is an essential TRIM/RBCC protein to maintain the integrity of sarcomeric cytoarchitecture. J. Cell Sci. 2013, 126, 3314–3323. [Google Scholar] [CrossRef] [Green Version]
- Bawa, S.; Brooks, D.S.; Neville, K.E.; Tipping, M.; Sagar, M.A.; Kollhoff, J.A.; Chawla, G.; Geisbrecht, B.V.; Tennessen, J.M.; Eliceiri, K.W.; et al. TRIM32 cooperates with glycolytic enzymes to promote cell growth. Elife 2020, 9. [Google Scholar] [CrossRef]
- LaBeau-DiMenna, E.M.; Clark, K.A.; Bauman, K.D.; Parker, D.S.; Cripps, R.M.; Geisbrecht, E.R. Thin, a Trim32 ortholog, is essential for myofibril stability and is required for the integrity of the costamere in Drosophila. Proc. Natl. Acad. Sci. USA 2012, 109, 17983–17988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borden, K.L.; Freemont, P.S. The RING finger domain: A recent example of a sequence-structure family. Curr. Opin. Struct. Biol. 1996, 6, 395–401. [Google Scholar] [CrossRef]
- Saurin, A.J.; Borden, K.L.; Boddy, M.N.; Freemont, P.S. Does this have a familiar RING? Trends Biochem. Sci. 1996, 21, 208–214. [Google Scholar] [CrossRef]
- Budhidarmo, R.; Nakatani, Y.; Day, C.L. RINGs hold the key to ubiquitin transfer. Trends Biochem. Sci. 2012, 37, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Tocchini, C.; Keusch, J.J.; Miller, S.B.; Finger, S.; Gut, H.; Stadler, M.B.; Ciosk, R. The TRIM-NHL protein LIN-41 controls the onset of developmental plasticity in Caenorhabditis elegans. PLoS Genet. 2014, 10, e1004533. [Google Scholar] [CrossRef] [Green Version]
- Koliopoulos, M.G.; Esposito, D.; Christodoulou, E.; Taylor, I.A.; Rittinger, K. Functional role of TRIM E3 ligase oligomerization and regulation of catalytic activity. EMBO J. 2016, 35, 1204–1218. [Google Scholar] [CrossRef]
- Massiah, M.A.; Simmons, B.N.; Short, K.M.; Cox, T.C. Solution structure of the RBCC/TRIM B-box1 domain of human MID1: B-box with a RING. J. Mol. Biol. 2006, 358, 532–545. [Google Scholar] [CrossRef]
- Lazzari, E.; El-Halawany, M.S.; De March, M.; Valentino, F.; Cantatore, F.; Migliore, C.; Onesti, S.; Meroni, G. Analysis of the Zn-Binding Domains of TRIM32, the E3 Ubiquitin Ligase Mutated in Limb Girdle Muscular Dystrophy 2H. Cells 2019, 8, 254. [Google Scholar] [CrossRef] [Green Version]
- Bawa, S.; Gameros, S.; Baumann, K.; Brooks, D.S.; Kollhoff, J.A.; Zolkiewski, M.; David Re Cecconi, A.; Panini, N.; Russo, M.; Piccirillo, R.; et al. Costameric Integrin and Sarcoglycan protein levels are altered in a Drosophila model for Limb Girdle Muscular Dystrophy type 2H. Mol. Biol. Cell 2020, mbcE20070453. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, M.; Sarcevic, B. Mechanisms of mono- and poly-ubiquitination: Ubiquitination specificity depends on compatibility between the E2 catalytic core and amino acid residues proximal to the lysine. Cell Div. 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissman, A.M. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2001, 2, 169–178. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtake, F.; Tsuchiya, H.; Saeki, Y.; Tanaka, K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. USA 2018, 115, E1401–E1408. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.V.; Soares, A.R.; Ramalho, J.S.; Pereira, P.; Girao, H. K63 linked ubiquitin chain formation is a signal for HIF1A degradation by Chaperone-Mediated Autophagy. Sci. Rep. 2015, 5, 10210. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albor, A.; El-Hizawi, S.; Horn, E.J.; Laederich, M.; Frosk, P.; Wrogemann, K.; Kulesz-Martin, M. The interaction of Piasy with Trim32, an E3-ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2H, promotes Piasy degradation and regulates UVB-induced keratinocyte apoptosis through NFkappaB. J. Biol. Chem. 2006, 281, 25850–25866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, M.; Tinsley, C.L.; Benson, M.A.; Blake, D.J. TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum. Mol. Genet. 2009, 18, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, E.; Kudryashov, D.; Kramerova, I.; Spencer, M.J. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 2005, 354, 413–424. [Google Scholar] [CrossRef]
- Kano, S.; Miyajima, N.; Fukuda, S.; Hatakeyama, S. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res. 2008, 68, 5572–5580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xu-Monette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Kaneko, Y. Trim32 facilitates degradation of MYCN on spindle poles and induces asymmetric cell division in human neuroblastoma cells. Cancer Res. 2014, 74, 5620–5630. [Google Scholar] [CrossRef] [Green Version]
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925. [Google Scholar] [CrossRef] [Green Version]
- Ryu, Y.S.; Lee, Y.; Lee, K.W.; Hwang, C.Y.; Maeng, J.S.; Kim, J.H.; Seo, Y.S.; You, K.H.; Song, B.; Kwon, K.S. TRIM32 protein sensitizes cells to tumor necrosis factor (TNFα)-induced apoptosis via its RING domain-dependent E3 ligase activity against X-linked inhibitor of apoptosis (XIAP). J. Biol. Chem. 2011, 286, 25729–25738. [Google Scholar] [CrossRef] [Green Version]
- Kudryashova, E.; Kramerova, I.; Spencer, M.J. Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. J. Clin. Investig. 2012, 122, 1764–1776. [Google Scholar] [CrossRef] [Green Version]
- Mokhonova, E.I.; Avliyakulov, N.K.; Kramerova, I.; Kudryashova, E.; Haykinson, M.J.; Spencer, M.J. The E3 ubiquitin ligase TRIM32 regulates myoblast proliferation by controlling turnover of NDRG2. Hum. Mol. Genet. 2015, 24, 2873–2883. [Google Scholar] [CrossRef] [Green Version]
- Overå, K.S.; Garcia-Garcia, J.; Bhujabal, Z.; Jain, A.; Øvervatn, A.; Larsen, K.B.; Deretic, V.; Johansen, T.; Lamark, T.; Sjøttem, E. TRIM32, but not its muscular dystrophy-associated mutant, positively regulates and is targeted to autophagic degradation by p62/SQSTM1. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Guglieri, M.; Straub, V.; Bushby, K.; Lochmüller, H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008, 21, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, O.A.; Jiang, X.M. Limb-girdle muscular dystrophies: Where next after six decades from the first proposal (Review). Mol. Med. Rep. 2014, 9, 1515–1532. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.P.; Straub, V. The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies. J. Neuromuscul. Dis 2015, 2, S7–S19. [Google Scholar] [CrossRef] [Green Version]
- Chiang, A.P.; Beck, J.S.; Yen, H.J.; Tayeh, M.K.; Scheetz, T.E.; Swiderski, R.E.; Nishimura, D.Y.; Braun, T.A.; Kim, K.Y.; Huang, J.; et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 2006, 103, 6287–6292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokeir, M.H.; Kobrinsky, N.L. Autosomal recessive muscular dystrophy in Manitoba Hutterites. Clin. Genet. 1976, 9, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Frosk, P.; Del Bigio, M.R.; Wrogemann, K.; Greenberg, C.R. Hutterite brothers both affected with two forms of limb girdle muscular dystrophy: LGMD2H and LGMD2I. Eur J. Hum. Genet. 2005, 13, 978–982. [Google Scholar] [CrossRef]
- Schoser, B.G.; Frosk, P.; Engel, A.G.; Klutzny, U.; Lochmüller, H.; Wrogemann, K. Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann. Neurol. 2005, 57, 591–595. [Google Scholar] [CrossRef]
- Servián-Morilla, E.; Cabrera-Serrano, M.; Rivas-Infante, E.; Carvajal, A.; Lamont, P.J.; Pelayo-Negro, A.L.; Ravenscroft, G.; Junckerstorff, R.; Dyke, J.M.; Fletcher, S.; et al. Altered myogenesis and premature senescence underlie human TRIM32-related myopathy. Acta Neuropathol. Commun. 2019, 7, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccone, V.; Palmieri, M.; Passamano, L.; Piluso, G.; Meroni, G.; Politano, L.; Nigro, V. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum. Mutat. 2008, 29, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Ruvkun, G. A novel repeat domain that is often associated with RING finger and B-box motifs. Trends Biochem. Sci. 1998, 23, 474–475. [Google Scholar] [CrossRef]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 2008, 283, 4069–4076. [Google Scholar] [CrossRef] [Green Version]
- Solaz-Fuster, M.C.; Gimeno-Alcañiz, J.V.; Ros, S.; Fernandez-Sanchez, M.E.; Garcia-Fojeda, B.; Criado Garcia, O.; Vilchez, D.; Dominguez, J.; Garcia-Rocha, M.; Sanchez-Piris, M.; et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 2008, 17, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Minassian, B.A.; Lee, J.R.; Herbrick, J.A.; Huizenga, J.; Soder, S.; Mungall, A.J.; Dunham, I.; Gardner, R.; Fong, C.Y.; Carpenter, S.; et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998, 20, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Serratosa, J.M.; Gómez-Garre, P.; Gallardo, M.E.; Anta, B.; de Bernabé, D.B.; Lindhout, D.; Augustijn, P.B.; Tassinari, C.A.; Malafosse, R.M.; Topcu, M.; et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum. Mol. Genet. 1999, 8, 345–352. [Google Scholar] [CrossRef]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef]
- Romá-Mateo, C.; Moreno, D.; Vernia, S.; Rubio, T.; Bridges, T.M.; Gentry, M.S.; Sanz, P. Lafora disease E3-ubiquitin ligase malin is related to TRIM32 at both the phylogenetic and functional level. BMC Evol. Biol. 2011, 11, 225. [Google Scholar] [CrossRef] [Green Version]
- Deol, K.K.; Lorenz, S.; Strieter, E.R. Enzymatic Logic of Ubiquitin Chain Assembly. Front. Physiol. 2019, 10, 835. [Google Scholar] [CrossRef]
- David, Y.; Ziv, T.; Admon, A.; Navon, A. The E2 ubiquitin-conjugating enzymes direct polyubiquitination to preferred lysines. J. Biol. Chem. 2010, 285, 8595–8604. [Google Scholar] [CrossRef] [Green Version]
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, E.; Struyk, A.; Mokhonova, E.; Cannon, S.C.; Spencer, M.J. The common missense mutation D489N in TRIM32 causing limb girdle muscular dystrophy 2H leads to loss of the mutated protein in knock-in mice resulting in a Trim32-null phenotype. Hum. Mol. Genet. 2011, 20, 3925–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the Muscle Cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaka, O.; Casas-Fraile, L.; López de Munain, A.; Sáenz, A. Costamere proteins and their involvement in myopathic processes. Expert Rev. Mol. Med. 2015, 17, e12. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.K.; Cheng, H.; Ross, R.S.; Knowlton, K.U.; Chen, J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog. Pediatr. Cardiol. 2011, 31, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Eid Mutlak, Y.; Aweida, D.; Volodin, A.; Ayalon, B.; Dahan, N.; Parnis, A.; Cohen, S. A signaling hub of insulin receptor, dystrophin glycoprotein complex and plakoglobin regulates muscle size. Nat. Commun. 2020, 11, 1381. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Lee, D.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglobin-PI3K dissociation. J. Cell Biol. 2014, 204, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Ebert, S.M.; Al-Zougbi, A.; Bodine, S.C.; Adams, C.M. Skeletal Muscle Atrophy: Discovery of Mechanisms and Potential Therapies. Physiology (Bethesda) 2019, 34, 232–239. [Google Scholar] [CrossRef]
- Piccirillo, R.; Demontis, F.; Perrimon, N.; Goldberg, A.L. Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev. Dyn. 2014, 243, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef]
- Barclay, R.D.; Burd, N.A.; Tyler, C.; Tillin, N.A.; Mackenzie, R.W. The Role of the IGF-1 Signaling Cascade in Muscle Protein Synthesis and Anabolic Resistance in Aging Skeletal Muscle. Front. Nutr. 2019, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Jamora, C.; Fuchs, E. Intercellular adhesion, signalling and the cytoskeleton. Nat. Cell Biol. 2002, 4, E101–E108. [Google Scholar] [CrossRef]
- Calautti, E.; Li, J.; Saoncella, S.; Brissette, J.L.; Goetinck, P.F. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J. Biol. Chem. 2005, 280, 32856–32865. [Google Scholar] [CrossRef] [Green Version]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Antonioli, M.; Fusco, C.; Liu, Y.; Mari, M.; Orhon, I.; Refolo, G.; Germani, F.; Corazzari, M.; Romagnoli, A.; et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci. Adv. 2019, 5, eaau8857. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Datzkiw, D.; Rudnicki, M.A. Satellite cells in ageing: Use it or lose it. Open Biol. 2020, 10, 200048. [Google Scholar] [CrossRef]
- Snijders, T.; Nederveen, J.P.; McKay, B.R.; Joanisse, S.; Verdijk, L.B.; van Loon, L.J.; Parise, G. Satellite cells in human skeletal muscle plasticity. Front. Physiol. 2015, 6, 283. [Google Scholar] [CrossRef] [Green Version]
- Assereto, S.; Piccirillo, R.; Baratto, S.; Scudieri, P.; Fiorillo, C.; Massacesi, M.; Traverso, M.; Galietta, L.J.; Bruno, C.; Minetti, C.; et al. The ubiquitin ligase tripartite-motif-protein 32 is induced in Duchenne muscular dystrophy. Lab. Investig. 2016, 96, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, S.; Otto, A.; Wu, X.; Miller, P.; Stelzer, S.; Wen, Y.; Kuang, S.; Wrogemann, K.; Patel, K.; Ding, H.; et al. TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PLoS ONE 2012, 7, e30445. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.B.; Gu, J.D.; Zhou, Q.H. Review of aerobic glycolysis and its key enzymes—new targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Green, H.J.; Helyar, R.; Ball-Burnett, M.; Kowalchuk, N.; Symon, S.; Farrance, B. Metabolic adaptations to training precede changes in muscle mitochondrial capacity. J. Appl. Physiol. (1985) 1992, 72, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Jang, H.C.; Lim, S. Differences among skeletal muscle mass indices derived from height-, weight-, and body mass index-adjusted models in assessing sarcopenia. Korean J. Intern. Med. 2016, 31, 643–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szent-Györgyi, A.G. The early history of the biochemistry of muscle contraction. J. Gen. Physiol. 2004, 123, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- Wojtas, K.; Slepecky, N.; von Kalm, L.; Sullivan, D. Flight muscle function in Drosophila requires colocalization of glycolytic enzymes. Mol. Biol. Cell 1997, 8, 1665–1675. [Google Scholar] [CrossRef]
- Sullivan, D.T.; MacIntyre, R.; Fuda, N.; Fiori, J.; Barrilla, J.; Ramizel, L. Analysis of glycolytic enzyme co-localization in Drosophila flight muscle. J. Exp. Biol. 2003, 206, 2031–2038. [Google Scholar] [CrossRef] [Green Version]
- Menard, L.; Maughan, D.; Vigoreaux, J. The structural and functional coordination of glycolytic enzymes in muscle: Evidence of a metabolon? Biology 2014, 3, 623–644. [Google Scholar] [CrossRef] [Green Version]
- Clarke, F.M.; Masters, C.J. On the association of glycolytic enzymes with structural proteins of skeletal muscle. Biochim Biophys. Acta 1975, 381, 37–46. [Google Scholar] [CrossRef]
- Shearwin, K.; Nanhua, C.; Masters, C. Interactions between glycolytic enzymes and cytoskeletal structure--the influence of ionic strength and molecular crowding. Biochem. Int. 1990, 21, 53–60. [Google Scholar]
- Waingeh, V.F.; Gustafson, C.D.; Kozliak, E.I.; Lowe, S.L.; Knull, H.R.; Thomasson, K.A. Glycolytic enzyme interactions with yeast and skeletal muscle F-actin. Biophys. J. 2006, 90, 1371–1384. [Google Scholar] [CrossRef] [Green Version]
- Araiza-Olivera, D.; Chiquete-Felix, N.; Rosas-Lemus, M.; Sampedro, J.G.; Peña, A.; Mujica, A.; Uribe-Carvajal, S. A glycolytic metabolon in Saccharomyces cerevisiae is stabilized by F-actin. FEBS J. 2013, 280, 3887–3905. [Google Scholar] [CrossRef]
- Tixier, V.; Bataillé, L.; Etard, C.; Jagla, T.; Weger, M.; Daponte, J.P.; Strähle, U.; Dickmeis, T.; Jagla, K. Glycolysis supports embryonic muscle growth by promoting myoblast fusion. Proc. Natl. Acad. Sci. USA 2013, 110, 18982–18987. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.M.; Gonzalez, M.; Poueymirou, W.T.; Kline, W.O.; Na, E.; Zlotchenko, E.; Stitt, T.N.; Economides, A.N.; Yancopoulos, G.D.; Glass, D.J. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol. Cell Biol. 2004, 24, 9295–9304. [Google Scholar] [CrossRef] [Green Version]
- Weitkunat, M.; Schnorrer, F. A guide to study Drosophila muscle biology. Methods 2014, 68, 2–14. [Google Scholar] [CrossRef]
- Demontis, F.; Perrimon, N. Integration of Insulin receptor/Foxo signaling and dMyc activity during muscle growth regulates body size in Drosophila. Development 2009, 136, 983–993. [Google Scholar] [CrossRef] [Green Version]
- Tennessen, J.M.; Baker, K.D.; Lam, G.; Evans, J.; Thummel, C.S. The Drosophila estrogen-related receptor directs a metabolic switch that supports developmental growth. Cell Metab. 2011, 13, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Rechsteiner, M.C. Drosophila lactate dehydrogenase and alpha-glycerolphosphate dehydrogenase: Distribution and change in activity during development. J. Insect. Physiol. 1970, 16, 1179–1192. [Google Scholar] [CrossRef]
- Abu-Shumays, R.L.; Fristrom, J.W. IMP-L3, A 20-hydroxyecdysone-responsive gene encodes Drosophila lactate dehydrogenase: Structural characterization and developmental studies. Dev. Genet. 1997, 20, 11–22. [Google Scholar] [CrossRef]
- Kim, M.J.; O’Connor, M.B. Activin signaling promotes muscle growth through InR/TORC1-dependent and -independent processes. Development 2021, 148. [Google Scholar] [CrossRef]
- Lauretani, F.; Russo, C.R.; Bandinelli, S.; Bartali, B.; Cavazzini, C.; Di Iorio, A.; Corsi, A.M.; Rantanen, T.; Guralnik, J.M.; Ferrucci, L. Age-associated changes in skeletal muscles and their effect on mobility: An operational diagnosis of sarcopenia. J. Appl. Physiol. (1985) 2003, 95, 1851–1860. [Google Scholar] [CrossRef]
- Gannon, J.; Staunton, L.; O’Connell, K.; Doran, P.; Ohlendieck, K. Phosphoproteomic analysis of aged skeletal muscle. Int. J. Mol. Med. 2008, 22, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef]
- Erez, A.; DeBerardinis, R.J. Metabolic dysregulation in monogenic disorders and cancer—Finding method in madness. Nat. Rev. Cancer 2015, 15, 440–448. [Google Scholar] [CrossRef]
- Heydemann, A. Skeletal Muscle Metabolism in Duchenne and Becker Muscular Dystrophy-Implications for Therapies. Nutrients 2018, 10, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, K.C.; Kong, S.W.; Walsh, R.J.; Salajegheh, M.; Moghadaszadeh, B.; Amato, A.A.; Nazareno, R.; Lin, Y.Y.; Krastins, B.; Sarracino, D.A.; et al. Fast-twitch sarcomeric and glycolytic enzyme protein loss in inclusion body myositis. Muscle. Nerve 2009, 39, 739–753. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yin, C.; Okamoto, H.; Jaffe, H.; Oldfield, E.H.; Zhuang, Z.; Vortmeyer, A.O.; Rushing, E.J. Proteomic analysis of inclusion body myositis. J. Neuropathol. Exp. Neurol. 2006, 65, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Tennessen, J.M.; Bertagnolli, N.M.; Evans, J.; Sieber, M.H.; Cox, J.; Thummel, C.S. Coordinated metabolic transitions during Drosophila embryogenesis and the onset of aerobic glycolysis. G3 (Bethesda) 2014, 4, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Dizdar, L.; Jünemann, L.M.; Werner, T.A.; Verde, P.E.; Baldus, S.E.; Stoecklein, N.H.; Knoefel, W.T.; Krieg, A. Clinicopathological and functional implications of the inhibitor of apoptosis proteins survivin and XIAP in esophageal cancer. Oncol. Lett. 2018, 15, 3779–3789. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.H.; Ding, W.X.; Zhou, J.; He, J.Y.; Xu, Y.; Gambotto, A.A.; Rabinowich, H.; Fan, J.; Yin, X.M. Expression of X-linked inhibitor-of-apoptosis protein in hepatocellular carcinoma promotes metastasis and tumor recurrence. Hepatology 2008, 48, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.G.; van der Valk, P.; Span, S.W.; Jonker, J.M.; Postmus, P.E.; Kruyt, F.A.; Giaccone, G. Assessment of IAP (inhibitor of apoptosis) proteins as predictors of response to chemotherapy in advanced non-small-cell lung cancer patients. Ann. Oncol. 2001, 12, 799–805. [Google Scholar] [CrossRef]
- Seligson, D.B.; Hongo, F.; Huerta-Yepez, S.; Mizutani, Y.; Miki, T.; Yu, H.; Horvath, S.; Chia, D.; Goodglick, L.; Bonavida, B. Expression of X-linked inhibitor of apoptosis protein is a strong predictor of human prostate cancer recurrence. Clin. Cancer Res. 2007, 13, 6056–6063. [Google Scholar] [CrossRef] [Green Version]
- Yim, J.H.; Kim, W.G.; Jeon, M.J.; Han, J.M.; Kim, T.Y.; Yoon, J.H.; Hong, S.J.; Song, D.E.; Gong, G.; Shong, Y.K.; et al. Association between expression of X-linked inhibitor of apoptosis protein and the clinical outcome in a BRAF V600E-prevalent papillary thyroid cancer population. Thyroid 2014, 24, 689–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, Y.; Ji, R.; Gu, Q.; Zhao, X.; Sun, B. Prognostic value of the X-linked inhibitor of apoptosis protein for invasive ductal breast cancer with triple-negative phenotype. Hum. Pathol. 2010, 41, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Vishal, K.; Bawa, S.; Brooks, D.; Bauman, K.; Geisbrecht, E.R. Thin is required for cell death in the Drosophila abdominal muscles by targeting DIAP1. Cell Death Dis. 2018, 9, 740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.D.; Kwon, Y.T. Molecular mechanisms controlling asymmetric and symmetric self-renewal of cancer stem cells. J. Anal. Sci. Technol. 2015, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef]
- Horn, E.J.; Albor, A.; Liu, Y.; El-Hizawi, S.; Vanderbeek, G.E.; Babcock, M.; Bowden, G.T.; Hennings, H.; Lozano, G.; Weinberg, W.C.; et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 2004, 25, 157–167. [Google Scholar] [CrossRef]
- Cui, X.; Lin, Z.; Chen, Y.; Mao, X.; Ni, W.; Liu, J.; Zhou, H.; Shan, X.; Chen, L.; Lv, J.; et al. Upregulated TRIM32 correlates with enhanced cell proliferation and poor prognosis in hepatocellular carcinoma. Mol. Cell Biochem. 2016, 421, 127–137. [Google Scholar] [CrossRef]
- Ito, M.; Migita, K.; Matsumoto, S.; Wakatsuki, K.; Tanaka, T.; Kunishige, T.; Nakade, H.; Nakatani, M.; Nakajima, Y. Overexpression of E3 ubiquitin ligase tripartite motif 32 correlates with a poor prognosis in patients with gastric cancer. Oncol. Lett. 2017, 13, 3131–3138. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.T.; Jin, F.; Li, J.G.; Xu, Y.Y.; Dong, H.T.; Liu, Q.; Xing, P.; Zhu, G.L.; Xu, H.; Yin, S.C.; et al. TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-κB pathway. J. Cancer 2018, 9, 1349–1356. [Google Scholar] [CrossRef]
- Luo, Q.; Wu, X.; Nan, Y.; Chang, W.; Zhao, P.; Zhang, Y.; Su, D.; Liu, Z. TRIM32/USP11 Balances ARID1A Stability and the Oncogenic/Tumor-Suppressive Status of Squamous Cell Carcinoma. Cell Rep. 2020, 30, 98–111.e115. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Li, Z.; Chen, J.; Hu, X. Expression and the potential functions of TRIM32 in lung cancer tumorigenesis. J. Cell Biochem. 2019, 120, 5232–5243. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, R.; Giavazzi, R. Inactivating STAT3: Bad for tumor, good for muscle. Cell Cycle 2015, 14, 939–940. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Zhang, W.; Du, B.; Zang, S.; Wang, X.; Mao, X.; Hu, Z. TRIM32 overexpression improves chemoresistance through regulation of mitochondrial function in non-small-cell lung cancers. Onco Targets Ther. 2018, 11, 7841–7852. [Google Scholar] [CrossRef] [Green Version]
- Drummond-Barbosa, D.; Tennessen, J.M. Reclaiming Warburg: Using developmental biology to gain insight into human metabolic diseases. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.W.; Purkayastha, A.; Jones, K.T.; Thaker, S.K.; Banerjee, U. In vivo genetic dissection of tumor growth and the Warburg effect. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Y.; Qu, D.; Jiang, T.; Li, S. Osthole induces G2/M arrest and apoptosis in lung cancer A549 cells by modulating PI3K/Akt pathway. J. Exp. Clin. Cancer Res. 2011, 30, 33. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, A.; Fukuoka, J.; Shimizu, S.; Shilo, K.; Franks, T.J.; Hewitt, S.M.; Fujii, T.; Cordon-Cardo, C.; Jen, J.; Travis, W.D. Overexpression of phospho-eIF4E is associated with survival through AKT pathway in non-small cell lung cancer. Clin. Cancer Res. 2010, 16, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, E.; Baba, H.; Tokunaga, E.; Nakamura, T.; Ueda, N.; Futatsugi, M.; Mashino, K.; Yamamoto, M.; Ikebe, M.; Kakeji, Y.; et al. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. Int. J. Cancer 2005, 117, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qu, X.; Hou, K.; Zhang, Y.; Dong, Q.; Teng, Y.; Zhang, J.; Liu, Y. PI3K/Akt is involved in bufalin-induced apoptosis in gastric cancer cells. Anticancer Drugs 2009, 20, 59–64. [Google Scholar] [CrossRef]
- Wang, L.; Ouyang, F.; Liu, X.; Wu, S.; Wu, H.M.; Xu, Y.; Wang, B.; Zhu, J.; Xu, X.; Zhang, L. Overexpressed CISD2 has prognostic value in human gastric cancer and promotes gastric cancer cell proliferation and tumorigenesis via AKT signaling pathway. Oncotarget 2016, 7, 3791–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Fang, Y.; Liu, T. TRIM32 Promotes the Growth of Gastric Cancer Cells through Enhancing AKT Activity and Glucose Transportation. Biomed. Res. Int. 2020, 2020, 4027627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathiraja, T.N.; Thakkar, K.N.; Jiang, S.; Stratton, S.; Liu, Z.; Gagea, M.; Shi, X.; Shah, P.K.; Phan, L.; Lee, M.H.; et al. TRIM24 links glucose metabolism with transformation of human mammary epithelial cells. Oncogene 2015, 34, 2836–2845. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, V.; Bowen, T.S.; Werner, S.; Barthel, P.; Amberger, C.; Konzer, A.; Graumann, J.; Sehr, P.; Lewis, J.; Provaznik, J.; et al. Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J. Cachexia Sarcopenia Muscle 2019, 10, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.; Gußen, V.; Zozulya, S.; Cruz, A.; Moriscot, A.; Linke, A.; Labeit, S. Small-Molecule Chemical Knockdown of MuRF1 in Melanoma Bearing Mice Attenuates Tumor Cachexia Associated Myopathy. Cells 2020, 9, 2272. [Google Scholar] [CrossRef]
- D’Amico, F.; Mukhopadhyay, R.; Ovaa, H.; Mulder, M.P.C. Targeting TRIM Proteins: A quest towards drugging an emerging protein class. Chembiochem 2021. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bawa, S.; Piccirillo, R.; Geisbrecht, E.R. TRIM32: A Multifunctional Protein Involved in Muscle Homeostasis, Glucose Metabolism, and Tumorigenesis. Biomolecules 2021, 11, 408. https://doi.org/10.3390/biom11030408
Bawa S, Piccirillo R, Geisbrecht ER. TRIM32: A Multifunctional Protein Involved in Muscle Homeostasis, Glucose Metabolism, and Tumorigenesis. Biomolecules. 2021; 11(3):408. https://doi.org/10.3390/biom11030408
Chicago/Turabian StyleBawa, Simranjot, Rosanna Piccirillo, and Erika R. Geisbrecht. 2021. "TRIM32: A Multifunctional Protein Involved in Muscle Homeostasis, Glucose Metabolism, and Tumorigenesis" Biomolecules 11, no. 3: 408. https://doi.org/10.3390/biom11030408
APA StyleBawa, S., Piccirillo, R., & Geisbrecht, E. R. (2021). TRIM32: A Multifunctional Protein Involved in Muscle Homeostasis, Glucose Metabolism, and Tumorigenesis. Biomolecules, 11(3), 408. https://doi.org/10.3390/biom11030408