
Investigation of the Differences in Antithrombin to Heparin Binding among Antithrombin Budapest 3, Basel, and Padua Mutations by Biochemical and In Silico Methods


 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antithrombin Deficient Patients and Their Routine Laboratory Investigation
2.2. Mutation Analysis of Antithrombin Deficient Patients
2.3. In Vitro Expression of Wild Type and Mutant Antithrombins
2.4. Preparation of Antithrombin from In Vitro Expressed Recombinant Antithrombins and from Normal and ATBp3 Homozygous Plasma by Affinity Chromatography
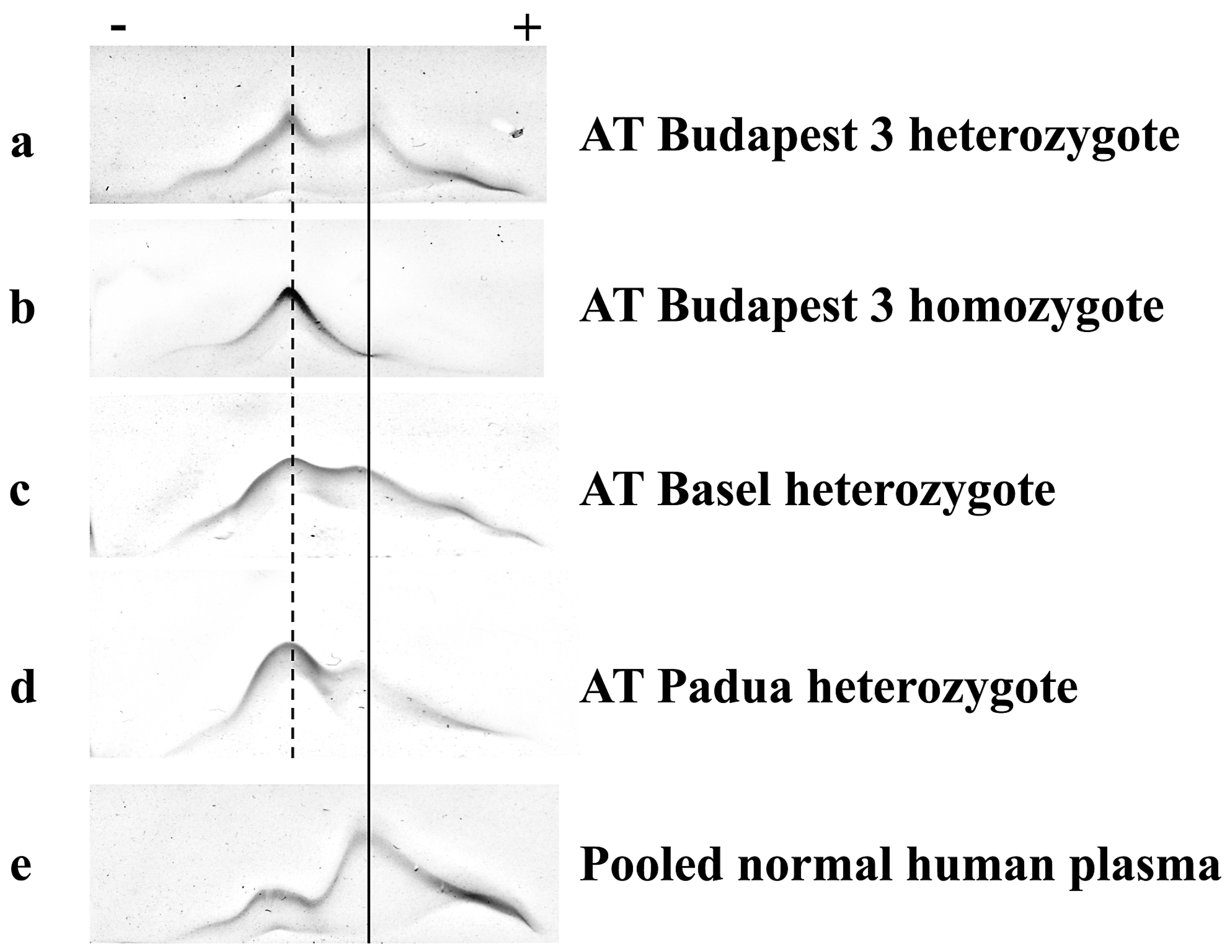
2.5. Crossed Immunoelectrophoresis
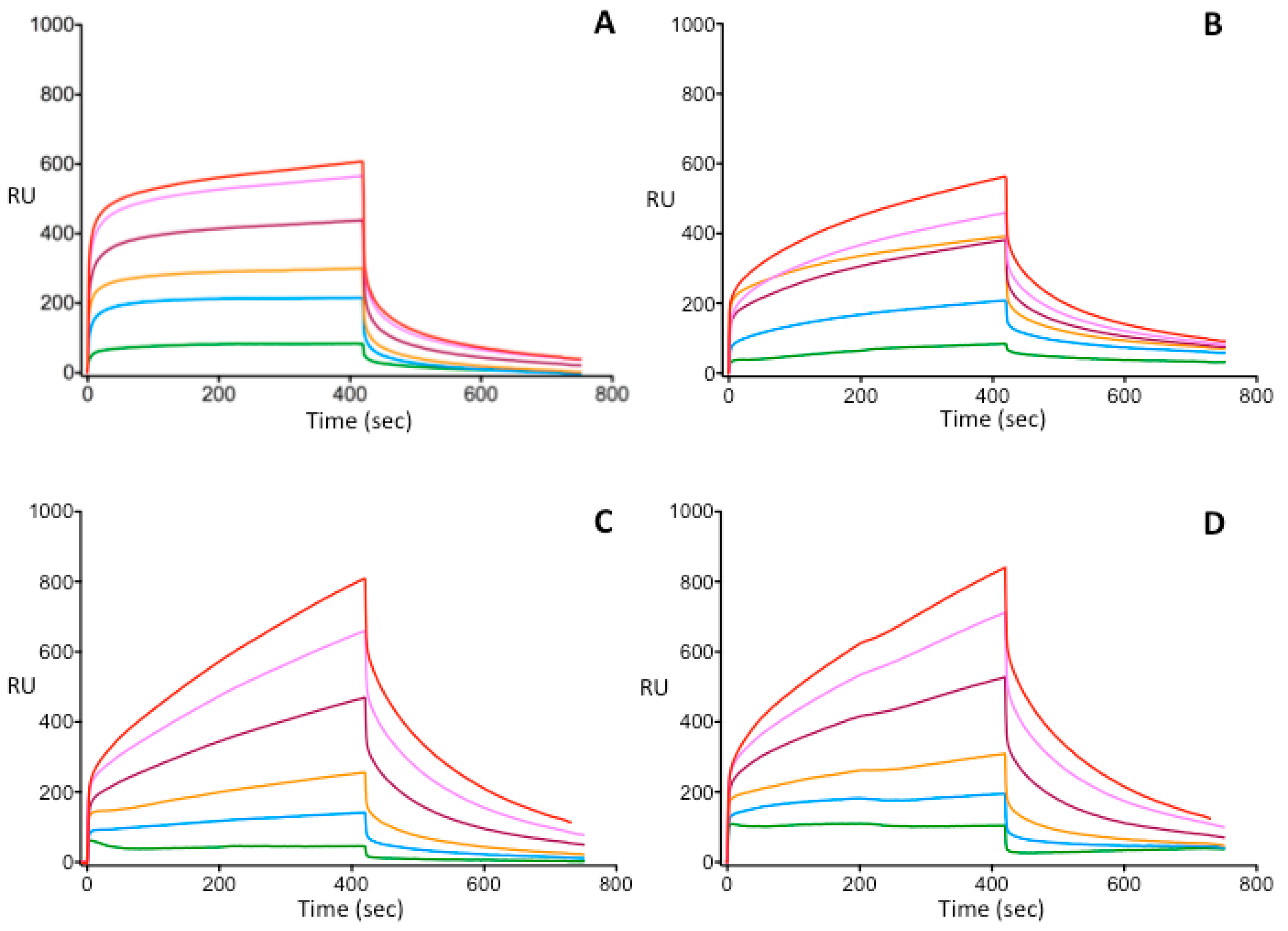
2.6. Surface Plasmon Resonance
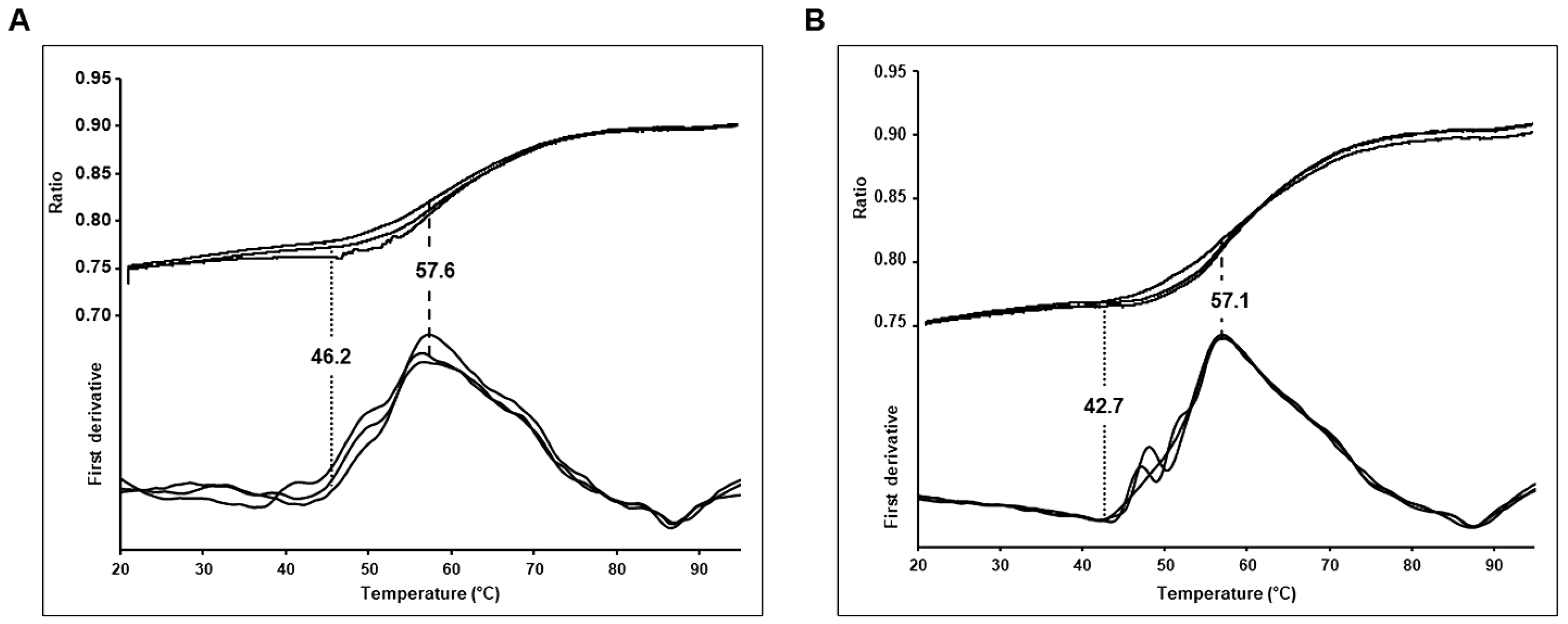
2.7. NanoDSF
2.8. In Silico Methods
3. Results
3.1. Clinical and Laboratory Characteristics of Patients with Antithrombin Type II Heparin-Binding Site Mutations
3.2. Thermostability of Wild Type and Budapest 3 Homozygous Antithrombin
3.3. Investigation of Heparin-Binding Characteristics of Wild Type and Different IIHBS Antithrombin Mutants
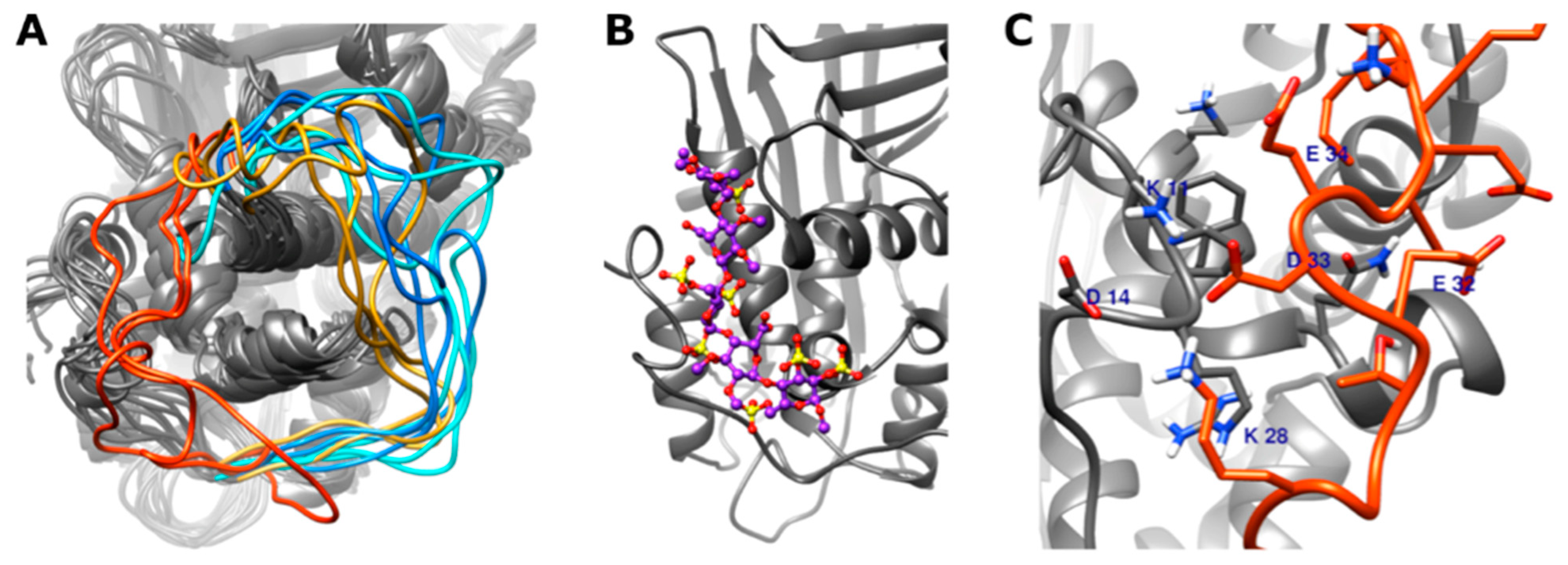
3.4. In Silico Modeling of Wild Type and Different Type IIHBS Mutant Antithrombin Proteins
3.4.1. Conformation of the 22–46 Loop
3.4.2. DSSP (Define Secondary Structure of Proteins) Analysis of the N-Terminal Part of the Antithrombin Protein
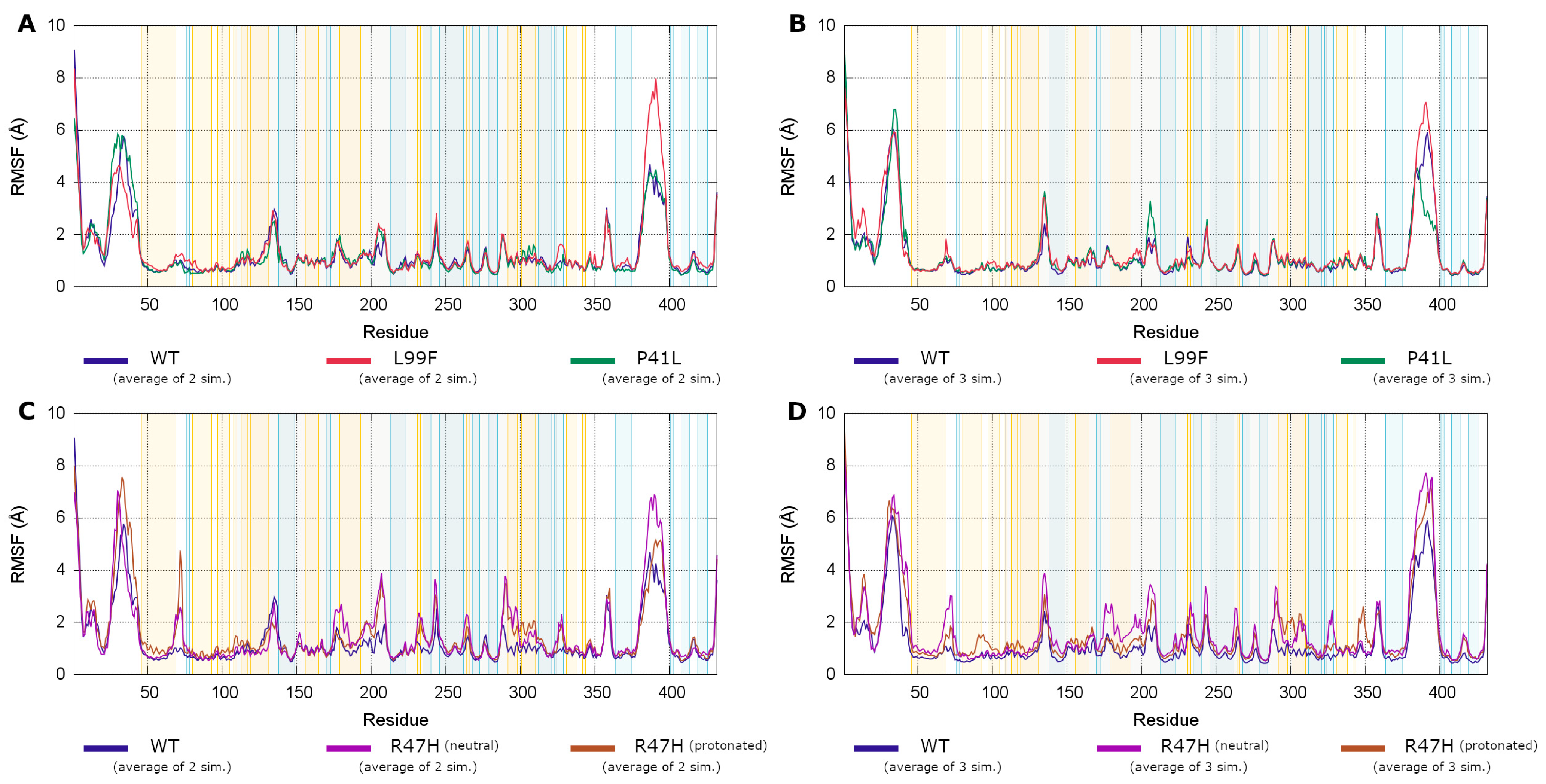
3.4.3. Root Mean Square Fluctuations Analysis
3.4.4. Analysis of Allosteric Pathways
3.4.5. Root Mean Square Deviations of Pentasaccharide Binding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muszbek, L.; Bereczky, Z.; Kovacs, B.; Komaromi, I. Antithrombin deficiency and its laboratory diagnosis. Clin. Chem. Lab. Med. 2010, 48 (Suppl. 1), S67–S78. [Google Scholar] [CrossRef]
- Egeberg, O. Inherited Antithrombin Deficiency Causing Thrombophilia. Thromb. Haemost. 1965, 13, 516–530. [Google Scholar] [CrossRef]
- Sas, G.; Blasko, G.; Banhegyi, D.; Jako, J.; Palos, L.A. Abnormal antithrombin III (antithrombin III “Budapest”) as a cause of a familial thrombophilia. Thromb. Haemost. 1974, 32, 105–115. [Google Scholar] [CrossRef]
- Lane, D.A.; Bayston, T.; Olds, R.J.; Fitches, A.C.; Cooper, D.N.; Millar, D.S.; Jochmans, K.; Perry, D.J.; Okajima, K.; Thein, S.L.; et al. Antithrombin mutation database: 2nd (1997) update. For the Plasma Coagulation Inhibitors Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost. 1997, 77, 197–211. [Google Scholar] [CrossRef]
- Lane, D.A.; Olds, R.J.; Conard, J.; Boisclair, M.; Bock, S.C.; Hultin, M.; Abildgaard, U.; Ireland, H.; Thompson, E.; Sas, G.; et al. Pleiotropic effects of antithrombin strand 1C substitution mutations. J. Clin. Investig. 1992, 90, 2422–2433. [Google Scholar] [CrossRef] [PubMed]
- Harmanci, O.; Ersoy, O.; Gurgey, A.; Buyukasik, Y.; Gedikoglu, G.; Balkanci, F.; Sivri, B.; Bayraktar, Y. The etiologic distribution of thrombophilic factors in chronic portal vein thrombosis. J. Clin. Gastroenterol. 2007, 41, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Ates, O. The deficiencies of protein C, protein S and antithrombin III in patients with retinal vein occlusion: A Turkish sample. Clin. Lab. Haematol. 2006, 28, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, D.; Martin, J.T.; Ferneini, A.M.; De Natale, R.W. Acute mesenteric and aortic thrombosis associated with antithrombin deficiency: A rare occurrence. Ann. Vasc. Surg. 2010, 24, 415.e5. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, K.; Tatsuta, K.; Orita, H.; Yonemura, T.; Kohno, H. Superior mesenteric and portal vein thrombosis caused by congenital antithrombin III deficiency: Report of a case. Surg. Today 2007, 37, 308–310. [Google Scholar] [CrossRef]
- Santangeli, P.; Sestito, A. Acute left atrial thrombosis during anticoagulant therapy in a patient with antithrombin deficiency. Acta Cardiol. 2008, 63, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.M.; Hsueg, C.H.; Chu, K.M.; Cheng, S.M.; Tsao, T.P. Simultaneous thromboses of double coronary arteries in a young male with antithrombin III deficiency. Am. J. Emerg. Med. 2009, 27, 1169.e3. [Google Scholar] [CrossRef]
- Peovska, I.; Maksimovic, J.; Kalpak, O.; Pejkov, H.; Bosevski, M. Recurrent myocardial infarction in a young football player with antithrombin III deficiency. Cardiol. J. 2008, 15, 463–466. [Google Scholar] [PubMed]
- Baiges, A.; de la Morena-Barrio, M.E.; Turon, F.; Minano, A.; Alberto Ferrusquia, J.; Magaz, M.; Reverter, J.C.; Vicente, V.; Hernandez-Gea, V.; Corral, J.; et al. Congenital antithrombin deficiency in patients with splanchnic vein thrombosis. Liver Int. 2020, 40, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Alhenc-Gelas, M.; Plu-Bureau, G.; Hugon-Rodin, J.; Picard, V.; Horellou, M.H. Thrombotic risk according to SERPINC1 genotype in a large cohort of subjects with antithrombin inherited deficiency. Thromb. Haemost. 2017, 117, 1040–1051. [Google Scholar] [CrossRef]
- Luxembourg, B.; Pavlova, A.; Geisen, C.; Spannagl, M.; Bergmann, F.; Krause, M.; Alesci, S.; Seifried, E.; Lindhoff-Last, E. Impact of the type of SERPINC1 mutation and subtype of antithrombin deficiency on the thrombotic phenotype in hereditary antithrombin deficiency. Thromb. Haemost. 2014, 111, 249–257. [Google Scholar] [CrossRef]
- Sekiya, A.; Taniguchi, F.; Yamaguchi, D.; Kamijima, S.; Kaneko, S.; Katsu, S.; Hanamura, M.; Takata, M.; Nakano, H.; Asakura, H.; et al. Causative genetic mutations for antithrombin deficiency and their clinical background among Japanese patients. Int. J. Hematol. 2017, 105, 287–294. [Google Scholar] [CrossRef]
- Kjaergaard, A.D.; Larsen, O.H.; Hvas, A.M.; Nissen, P.H. SERPINC1 variants causing hereditary antithrombin deficiency in a Danish population. Thromb. Res. 2019, 175, 68–75. [Google Scholar] [CrossRef]
- Wang, D.; Cui, G.; Hu, S.; Wang, D.W. Subtypes of SERPINC1 mutations and the thrombotic phenotype of inherited antithrombin deficient individuals in Chinese Han population. Blood Cells Mol. Dis. 2016, 62, 38–41. [Google Scholar] [CrossRef]
- Puurunen, M.; Salo, P.; Engelbarth, S.; Javela, K.; Perola, M. Type II antithrombin deficiency caused by a founder mutation Pro73Leu in the Finnish population: Clinical picture. J. Thromb. Haemost. 2013, 11, 1844–1849. [Google Scholar] [CrossRef]
- Orlando, C.; Heylen, O.; Lissens, W.; Jochmans, K. Antithrombin heparin binding site deficiency: A challenging diagnosis of a not so benign thrombophilia. Thromb. Res. 2015, 135, 1179–1185. [Google Scholar] [CrossRef]
- Gindele, R.; Selmeczi, A.; Olah, Z.; Ilonczai, P.; Pfliegler, G.; Marjan, E.; Nemes, L.; Nagy, A.; Losonczy, H.; Mitic, G.; et al. Clinical and laboratory characteristics of antithrombin deficiencies: A large cohort study from a single diagnostic center. Thromb. Res. 2017, 160, 119–128. [Google Scholar] [CrossRef]
- Gindele, R.; Olah, Z.; Ilonczai, P.; Speker, M.; Udvari, A.; Selmeczi, A.; Pfliegler, G.; Marjan, E.; Kovacs, B.; Boda, Z.; et al. Founder effect is responsible for the p.Leu131Phe heparin-binding-site antithrombin mutation common in Hungary: Phenotype analysis in a large cohort. J. Thromb. Haemost. 2016, 14, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bereczky, Z.; Gindele, R.; Fiatal, S.; Speker, M.; Miklos, T.; Balogh, L.; Mezei, Z.; Szabo, Z.; Adany, R. Age and Origin of the Founder Antithrombin Budapest 3 (p.Leu131Phe) Mutation; Its High Prevalence in the Roma Population and Its Association With Cardiovascular Diseases. Front. Cardiovasc. Med. 2020, 7, 617711. [Google Scholar] [CrossRef]
- Perry, D.J.; Daly, M.E.; Tait, R.C.; Walker, I.D.; Brown, K.; Beauchamp, N.J.; Preston, F.E.; Gyde, H.; Harper, P.L.; Carrell, R.W. Antithrombin cambridge II (Ala384Ser): Clinical, functional and haplotype analysis of 18 families. Thromb. Haemost. 1998, 79, 249–253. [Google Scholar]
- Corral, J.; Hernandez-Espinosa, D.; Soria, J.M.; Gonzalez-Conejero, R.; Ordonez, A.; Gonzalez-Porras, J.R.; Perez-Ceballos, E.; Lecumberri, R.; Sanchez, I.; Roldan, V.; et al. Antithrombin Cambridge II (A384S): An underestimated genetic risk factor for venous thrombosis. Blood 2007, 109, 4258–4263. [Google Scholar] [CrossRef]
- Kovacs, B.; Bereczky, Z.; Olah, Z.; Gindele, R.; Kerenyi, A.; Selmeczi, A.; Boda, Z.; Muszbek, L. The superiority of anti-FXa assay over anti-FIIa assay in detecting heparin-binding site antithrombin deficiency. Am. J. Clin. Pathol. 2013, 140, 675–679. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, B.; Bereczky, Z.; Selmeczi, A.; Gindele, R.; Olah, Z.; Kerenyi, A.; Boda, Z.; Muszbek, L. Progressive chromogenic anti-factor Xa assay and its use in the classification of antithrombin deficiencies. Clin. Chem. Lab. Med. 2014, 52, 1797–1806. [Google Scholar] [CrossRef]
- Bereczky, Z.; Gindele, R.; Speker, M.; Kallai, J. Deficiencies of the Natural Anticoagulants—Novel Clinical Laboratory Aspects of Thrombophilia Testing. EJIFCC 2016, 27, 130–146. [Google Scholar]
- Selmeczi, A.; Gindele, R.; Ilonczai, P.; Fekete, A.; Komaromi, I.; Schlammadinger, A.; Razso, K.; Kovacs, K.B.; Bardos, H.; Adany, R.; et al. Antithrombin Debrecen (p.Leu205Pro)—Clinical and molecular characterization of a novel mutation associated with severe thrombotic tendency. Thromb. Res. 2017, 158, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Dinarvand, P.; Yang, L.; Villoutreix, B.O.; Rezaie, A.R. Expression and functional characterization of two natural heparin-binding site variants of antithrombin. J. Thromb. Haemost. 2018, 16, 330–341. [Google Scholar] [CrossRef]
- Sas, G.; Pepper, D.S.; Cash, J.D. Investigations on antithrombin III in normal plasma and serum. Br. J. Haematol. 1975, 30, 265–272. [Google Scholar] [CrossRef]
- Johnson, D.J.; Langdown, J.; Li, W.; Luis, S.A.; Baglin, T.P.; Huntington, J.A. Crystal structure of monomeric native antithrombin reveals a novel reactive center loop conformation. J. Biol. Chem. 2006, 281, 35478–35486. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.J.; Huntington, J.A. Crystal structure of antithrombin in a heparin-bound intermediate state. Biochemistry 2003, 42, 8712–8719. [Google Scholar] [CrossRef]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W., 2nd; Mackerell, A.D., Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [Green Version]
- Mallajosyula, S.S.; Guvench, O.; Hatcher, E.; Mackerell, A.D., Jr. CHARMM Additive All-Atom Force Field for Phosphate and Sulfate Linked to Carbohydrates. J. Chem. Theory Comput. 2012, 8, 759–776. [Google Scholar] [CrossRef] [Green Version]
- Brennan, S.O.; George, P.M.; Jordan, R.E. Physiological variant of antithrombin-III lacks carbohydrate sidechain at Asn 135. FEBS Lett. 1987, 219, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.J.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, J.; Patel, D.S.; Ma, H.; Lee, H.S.; Jo, S.; Im, W. Glycan Reader is improved to recognize most sugar types and chemical modifications in the Protein Data Bank. Bioinformatics 2017, 33, 3051–3057. [Google Scholar] [CrossRef]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- York, D.M.; Wlodawer, A.; Pedersen, L.G.; Darden, T.A. Atomic-level accuracy in simulations of large protein crystals. Proc. Natl. Acad. Sci. USA 1994, 91, 8715–8718. [Google Scholar] [CrossRef] [Green Version]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 1991, 11, 205–217. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Girolami, A.; Fabris, F.; Cappellato, G.; Sainati, L.; Boeri, G. Antithrombin III (AT III) Padua2: A “new” congenital abnormality with defective heparin co-factor activities but no thrombotic disease. Blut 1983, 47, 93–103. [Google Scholar] [CrossRef]
- Fitton, H.L.; Skinner, R.; Dafforn, T.R.; Jin, L.; Pike, R.N. The N-terminal segment of antithrombin acts as a steric gate for the binding of heparin. Protein Sci. 1998, 7, 782–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, G.; Komaromi, I.; Bereczky, Z. The mechanism of high affinity pentasaccharide binding to antithrombin, insights from Gaussian accelerated molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 38, 4718–4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, O.F.; Grubmuller, H. Generalized correlation for biomolecular dynamics. Proteins 2006, 62, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.Y.; Tran, T.H. Antithrombin III Basel. Identification of a Pro-Leu substitution in a hereditary abnormal antithrombin with impaired heparin cofactor activity. J. Biol. Chem. 1986, 261, 1174–1176. [Google Scholar] [CrossRef]
- Brennan, S.O.; Borg, J.Y.; Shaw, G.J.; Caen, J.; George, P.M. Functional antithrombin-III variant (41 Pro----Leu) identified by liquid secondary ion mass spectrometry. Pathology 1991, 23, 1–4. [Google Scholar] [CrossRef]
- Chowdhury, V.; Mille, B.; Olds, R.J.; Lane, D.A.; Watton, J.; Barrowcliffe, T.W.; Pabinger, I.; Woodcock, B.E.; Thein, S.L. Antithrombins Southport (Leu 99 to Val) and Vienna (Gln 118 to Pro): Two novel antithrombin variants with abnormal heparin binding. Br. J. Haematol. 1995, 89, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Arocas, V.; Bock, S.C.; Olson, S.T.; Bjork, I. The role of Arg46 and Arg47 of antithrombin in heparin binding. Biochemistry 1999, 38, 10196–10204. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, I.; Navarro-Fernandez, J.; Ostergaard, A.; Gutierrez-Gallego, R.; Padilla, J.; Bohdan, N.; Minano, A.; Pascual, C.; Martinez, C.; de la Morena-Barrio, M.E.; et al. Amelioration of the severity of heparin-binding antithrombin mutations by posttranslational mosaicism. Blood 2012, 120, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Olds, R.J.; Lane, D.A.; Boisclair, M.; Sas, G.; Bock, S.C.; Thein, S.L. Antithrombin Budapest 3. An antithrombin variant with reduced heparin affinity resulting from the substitution L99F. FEBS Lett. 1992, 300, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Kraft, J.; Sunder-Plassmann, R.; Mannhalter, C.; Quehenberger, P.; Tews, G.; Langer, M.; Pabinger, I. Women with homozygous AT deficiency type II heparin-binding site (HBS) are at high risk of pregnancy loss and pregnancy complications. Ann. Hematol. 2017, 96, 1023–1031. [Google Scholar] [CrossRef]
- Vossen, C.Y.; Conard, J.; Fontcuberta, J.; Makris, M.; FJ, V.D.M.; Pabinger, I.; Palareti, G.; Preston, F.E.; Scharrer, I.; Souto, J.C.; et al. Risk of a first venous thrombotic event in carriers of a familial thrombophilic defect. The European Prospective Cohort on Thrombophilia (EPCOT). J. Thromb. Haemost. 2005, 3, 459–464. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Ueda, K.; Ozawa, T.; Sakuragawa, N.; Yokota, S.; Sato, R.; Okamura, S.; Morita, M.; Imanaka, T. Intracellular accumulation of antithrombin Morioka (C95R), a novel mutation causing type I antithrombin deficiency. J. Biol. Chem. 2002, 277, 51058–51067. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.F.; Fu, Q.H.; Wang, W.B.; Xie, S.; Dai, J.; Ding, Q.L.; Wang, X.F.; Wang, H.L.; Wang, Z.Y. Molecular mechanisms of antithrombin deficiency in two Chinese families. One novel and one recurrent point mutation in the antithrombin gene causing venous thrombosis. Thromb. Haemost. 2005, 94, 1172–1176. [Google Scholar] [CrossRef] [Green Version]
- Homola, J. Surface plasmon resonance sensors for detection of chemical and biological species. Chem. Rev. 2008, 108, 462–493. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Malhotra, A.; Li, Q.; Zhang, F.; Linhardt, R.J. Novel method for measurement of heparin anticoagulant activity using SPR. Anal. BioChem. 2017, 526, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Z.; Lin, X.; Beenken, A.; Eliseenkova, A.V.; Mohammadi, M.; Linhardt, R.J. Compositional analysis of heparin/heparan sulfate interacting with fibroblast growth factor.fibroblast growth factor receptor complexes. Biochemistry 2009, 48, 8379–8386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Aguilera, J.; Beaudet, J.M.; Xie, Q.; Lerch, T.F.; Davulcu, O.; Colon, W.; Chapman, M.S.; Linhardt, R.J. Characterization of interactions between heparin/glycosaminoglycan and adeno-associated virus. Biochemistry 2013, 52, 6275–6285. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liu, X.; Kao, C.; Zhang, E.; Li, Q.; Zhang, F.; Linhardt, R.J. Kinetic and Structural Studies of Interactions between Glycosaminoglycans and Langerin. Biochemistry 2016, 55, 4552–4559. [Google Scholar] [CrossRef]
- Mirow, N.; Zimmermann, B.; Maleszka, A.; Knobl, H.; Tenderich, G.; Koerfer, R.; Herberg, F.W. Plasma protein binding properties to immobilized heparin and heparin-albumin conjugate. Artif. Organs 2007, 31, 466–471. [Google Scholar] [CrossRef]
- Miao, Y.; Bhattarai, A.; Wang, J. Ligand Gaussian Accelerated Molecular Dynamics (LiGaMD): Characterization of Ligand Binding Thermodynamics and Kinetics. J. Chem. Theory Comput. 2020, 16, 5526–5547. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AT Basel | AT Padua | ATBp3 Homozygous | ATBp3 Heterozygous | p Value | |
|---|---|---|---|---|---|
| n = 19 | n = 31 | n = 52 | n = 239 | ||
| Male/female | 6/13 | 8/23 | 25/27 | 98/141 | NS |
| Heparin cofactor anti-FXa AT activity (%) | 58 (44–74) | 57 (40–70) | 17 (9–53) + | 57 (36–86) | <0.001 |
| Progressive anti-FXa AT activity (%) | 100 (73–120) | 106 (73–126) | 73 (56–100) + | 85 (60–228) ++ | <0.001 |
| p-anti-FXa to hc-anti-FXa ratio | 1.77 (1.25–2.05) | 1.84 (1.28–2.10) | 4.30 (2.06–8.40) + | 1.50 (0.94–3.12) ++ | <0.001 |
| AT antigen (g/L) | 0.30 (0.25–0.36) | 0.30 (0.24–0.35) | 0.21 (0.13–0.29) + | 0.24 (0.14–0.35) ++ | <0.001 |
| Frequency of patients with VTE % | 11.1 | 26.9 | 88.8 § | 42.1 | <0.001 |
| Frequency of patients with PE % | 0 | 11.5 | 6.7 | 6.3 | NS |
| Frequency of patients with ATE % | 44.4 §§ | 7.7 | 6.7 | 7.6 | 0.002 |
| Frequency of females with pregnancy complications % | 37.5 | 42.9 | 38.9 | 35.4 | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gindele, R.; Pénzes-Daku, K.; Balogh, G.; Kállai, J.; Bogáti, R.; Bécsi, B.; Erdődi, F.; Katona, É.; Bereczky, Z. Investigation of the Differences in Antithrombin to Heparin Binding among Antithrombin Budapest 3, Basel, and Padua Mutations by Biochemical and In Silico Methods. Biomolecules 2021, 11, 544. https://doi.org/10.3390/biom11040544
Gindele R, Pénzes-Daku K, Balogh G, Kállai J, Bogáti R, Bécsi B, Erdődi F, Katona É, Bereczky Z. Investigation of the Differences in Antithrombin to Heparin Binding among Antithrombin Budapest 3, Basel, and Padua Mutations by Biochemical and In Silico Methods. Biomolecules. 2021; 11(4):544. https://doi.org/10.3390/biom11040544
Chicago/Turabian StyleGindele, Réka, Krisztina Pénzes-Daku, Gábor Balogh, Judit Kállai, Réka Bogáti, Bálint Bécsi, Ferenc Erdődi, Éva Katona, and Zsuzsanna Bereczky. 2021. "Investigation of the Differences in Antithrombin to Heparin Binding among Antithrombin Budapest 3, Basel, and Padua Mutations by Biochemical and In Silico Methods" Biomolecules 11, no. 4: 544. https://doi.org/10.3390/biom11040544
APA StyleGindele, R., Pénzes-Daku, K., Balogh, G., Kállai, J., Bogáti, R., Bécsi, B., Erdődi, F., Katona, É., & Bereczky, Z. (2021). Investigation of the Differences in Antithrombin to Heparin Binding among Antithrombin Budapest 3, Basel, and Padua Mutations by Biochemical and In Silico Methods. Biomolecules, 11(4), 544. https://doi.org/10.3390/biom11040544