
Cluster Analysis of Medicinal Plants and Targets Based on Multipartite Network


Abstract
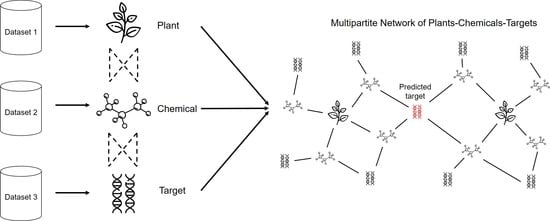
:
1. Introduction
2. Materials and Methods
2.1. Raw Data Sets
2.2. Preprocessing
2.3. Methods
3. Results
3.1. Multipartite Network Analysis
3.2. Hierarchical Clustering Analysis of Plants
3.3. Interaction between Plants and Targets Based on TPS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Petrovska, B. Historical Review of Medicinal Plants’ Usage. Pharmacogn. Rev. 2012, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Porras, G.; Chassagne, F.; Lyles, J.T.; Marquez, L.; Dettweiler, M.; Salam, A.M.; Samarakoon, T.; Shabih, S.; Farrokhi, D.R.; Quave, C.L. Ethnobotany and the Role of Plant Natural Products in Antibiotic Drug Discovery. Chem. Rev. 2021, 121, 3495–3560. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The Re-Emergence of Natural Products for Drug Discovery in the Genomics Era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehn, F.E.; Carter, G.T. The Evolving Role of Natural Products in Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on Natural Products for Drug Design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Galanakis, C.M. Nutraceuticals and Natural Product Pharmaceuticals; Academic Press: Cambridge, MA, USA, 2019; ISBN 978-0-12-816450-1. [Google Scholar]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery: Miniperspective. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Besnard, J.; Ruda, G.F.; Setola, V.; Abecassis, K.; Rodriguiz, R.M.; Huang, X.-P.; Norval, S.; Sassano, M.F.; Shin, A.I.; Webster, L.A.; et al. Automated Design of Ligands to Polypharmacological Profiles. Nature 2012, 492, 215–220. [Google Scholar] [CrossRef]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting New Molecular Targets for Known Drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S.; et al. Large-Scale Prediction and Testing of Drug Activity on Side-Effect Targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Trans. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L.; Sheffler, D.J.; Kroeze, W.K. Magic Shotguns versus Magic Bullets: Selectively Non-Selective Drugs for Mood Disorders and Schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Hong, H.; Mendrick, D.L.; Tang, Y.; Cheng, F. Biomarker-Based Drug Safety Assessment in the Age of Systems Pharmacology: From Foundational to Regulatory Science. Biomark. Med. 2015, 9, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Li, X.; Peng, S.; Wang, S.; Huang, C.Z.; Huang, C.Z.; Zhang, Q.; Li, D.; Jiang, J.; et al. Identification of a Specific Agonist of Human TAS2R14 from Radix Bupleuri through Virtual Screening, Functional Evaluation and Binding Studies. Sci. Rep. 2017, 7, 12174. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.; Yang, J.; Karan, C.; Menden, M.P.; Costello, J.C.; Tang, H.; Xiao, G.; Li, Y.; Allen, J.; Zhong, R.; et al. A Community Computational Challenge to Predict the Activity of Pairs of Compounds. Nat. Biotechnol. 2014, 32, 1213–1222. [Google Scholar] [CrossRef]
- Han, K.; Jeng, E.E.; Hess, G.T.; Morgens, D.W.; Li, A.; Bassik, M.C. Synergistic Drug Combinations for Cancer Identified in a CRISPR Screen for Pairwise Genetic Interactions. Nat. Biotechnol. 2017, 35, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhu, F.; Ma, X.; Cao, Z.W.; Li, Y.X.; Chen, Y.Z. Mechanisms of Drug Combinations: Interaction and Network Perspectives. Nat. Rev. Drug Discov. 2009, 8, 111–128. [Google Scholar] [CrossRef]
- Sun, Y.; Sheng, Z.; Ma, C.; Tang, K.; Zhu, R.; Wu, Z.; Shen, R.; Feng, J.; Wu, D.; Huang, D.; et al. Combining Genomic and Network Characteristics for Extended Capability in Predicting Synergistic Drugs for Cancer. Nat. Commun. 2015, 6, 8481. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K. The Worldwide Trend of Using Botanical Drugs and Strategies for Developing Global Drugs. BMB Rep. 2017, 50, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Ezzat, A.; Wu, M.; Li, X.; Kwoh, C.-K. Computational Prediction of Drug-Target Interactions via Ensemble Learning. In Computational Methods for Drug Repurposing; Vanhaelen, Q., Ed.; Springer: New York, NY, USA, 2019; Volume 1903, pp. 239–254. ISBN 978-1-4939-8954-6. [Google Scholar]
- Forouzesh, A.; Samadi Foroushani, S.; Forouzesh, F.; Zand, E. Reliable Target Prediction of Bioactive Molecules Based on Chemical Similarity Without Employing Statistical Methods. Front. Pharmacol. 2019, 10, 835. [Google Scholar] [CrossRef] [Green Version]
- Lavecchia, A.; Cerchia, C. In Silico Methods to Address Polypharmacology: Current Status, Applications and Future Perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A Network Integration Approach for Drug-Target Interaction Prediction and Computational Drug Repositioning from Heterogeneous Information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.L. Network Pharmacology: The next Paradigm in Drug Discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, N.R.C.; Ribeiro, B.; Arrais, J. Drug-Target Interaction Prediction: End-to-End Deep Learning Approach. IEEE/ACM Trans. Comput Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Cheng, F.; Kovács, I.A.; Barabási, A.-L. Network-Based Prediction of Drug Combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Leung, E.L.; Cao, Z.-W.; Jiang, Z.-H.; Zhou, H.; Liu, L. Network-Based Drug Discovery by Integrating Systems Biology and Computational Technologies. Brief. Bioinform. 2013, 14, 491–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yıldırım, M.A.; Goh, K.-I.; Cusick, M.E.; Barabási, A.-L.; Vidal, M. Drug—Target Network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jiang, P.; Zhang, W. Molecular Networks for the Study of TCM Pharmacology. Brief. Bioinform. 2010, 11, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Bosc, N.; Atkinson, F.; Felix, E.; Gaulton, A.; Hersey, A.; Leach, A.R. Large Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug Discovery. J. Cheminform. 2019, 11. [Google Scholar] [CrossRef]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Koutsoukas, A.; Simms, B.; Kirchmair, J.; Bond, P.J.; Whitmore, A.V.; Zimmer, S.; Young, M.P.; Jenkins, J.L.; Glick, M.; Glen, R.C.; et al. From in Silico Target Prediction to Multi-Target Drug Design: Current Databases, Methods and Applications. J. Proteom. 2011, 74, 2554–2574. [Google Scholar] [CrossRef]
- Shin, J.-S.; Lee, Y.-S.; Lee, M.-S. Protection and Utilization of Traditional Knowledge Resources through Korean Traditional Knowledge Portal(KTKP). J. Korea Contents Assoc. 2010, 10, 422–426. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, I.S. Co-Clustering Documents and Words Using Bipartite Spectral Graph Partitioning. In Proceedings of the Seventh ACM SIGKDD International Conference on Knowledge Discovery and Data Mining—KDD ’01, San Francisco, CA, USA, 26–29 August 2001; ACM Press: San Francisco, CA, USA, 2001; pp. 269–274. [Google Scholar]
- Martínez-Jiménez, F.; Papadatos, G.; Yang, L.; Wallace, I.M.; Kumar, V.; Pieper, U.; Sali, A.; Brown, J.R.; Overington, J.P.; Marti-Renom, M.A. Target Prediction for an Open Access Set of Compounds Active against Mycobacterium Tuberculosis. PLoS Comput. Biol. 2013, 9, e1003253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.E.J. Modularity and Community Structure in Networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.E.J. Fast Algorithm for Detecting Community Structure in Networks. Phys. Rev. E 2004, 69, 066133. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.E.J.; Girvan, M. Finding and Evaluating Community Structure in Networks. Phys. Rev. E 2004, 69, 026113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, K.-I.; Oh, E.; Jeong, H.; Kahng, B.; Kim, D. Classification of Scale-Free Networks. Proc. Natl. Acad. Sci. USA 2002, 99, 12583–12588. [Google Scholar] [CrossRef] [Green Version]
- Lima-Mendez, G.; van Helden, J. The Powerful Law of the Power Law and Other Myths in Network Biology. Mol. BioSyst. 2009, 5, 1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M. Power Laws, Pareto Distributions and Zipf’s Law. Contemp. Phys. 2005, 46, 323–351. [Google Scholar] [CrossRef] [Green Version]
- Tao, C.; Shkumatov, A.A.; Alexander, S.T.; Ason, B.L.; Zhou, M. Stigmasterol Accumulation Causes Cardiac Injury and Promotes Mortality. Commun. Biol. 2019, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, K.; Xi, Y.; Zhu, B.; Gary, B.D.; Ramírez-Alcántara, V.; Gurpinar, E.; Canzoneri, J.C.; Fajardo, A.; Sigler, S.; et al. Phosphodiesterase 10A: A Novel Target for Selective Inhibition of Colon Tumor Cell Growth and β-Catenin-Dependent TCF Transcriptional Activity. Oncogene 2015, 34, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Wu, J. Cannabis, Cannabinoid Receptors, and Endocannabinoid System: Yesterday, Today, and Tomorrow. Acta Pharmacol. Sin. 2019, 40, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Zhang, K.; Jin, Y.; Li, B.; Gao, S.; Zhu, J.; Cui, R. Pharmacological Effects of Berberine on Mood Disorders. J. Cell. Mol. Med. 2019, 23, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Takagi, R.; Kaneko, A.; Matsushita, S. Berberine Is a Dopamine D1- and D2-like Receptor Antagonist and Ameliorates Experimentally Induced Colitis by Suppressing Innate and Adaptive Immune Responses. J. Neuroimmunol. 2015, 289, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, G.; Kasiri, Y.; Birta, E.; Béke, G.; Garcia, A.L.; Bartók, E.-M.; Rühl, R. Lycopene-Derived Bioactive Retinoic Acid Receptors/Retinoid-X Receptors-Activating Metabolites May Be Relevant for Lycopene’s Anti-Cancer Potential. Mol. Nutr. Food Res. 2013, 57, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, G.; Carlsen, H.; Blomhoff, R.; Rühl, R. Lycopene Induces Retinoic Acid Receptor Transcriptional Activation in Mice. Mol. Nutr. Food Res. 2012, 56, 702–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Gao, Y.; Zhi, X.; Ta, N.; Jiang, H.; Zheng, J. Association between Vitamin A, Retinol and Carotenoid Intake and Pancreatic Cancer Risk: Evidence from Epidemiologic Studies. Sci. Rep. 2016, 6, 38936. [Google Scholar] [CrossRef]
- Smit, N.; Vicanova, J.; Pavel, S. The Hunt for Natural Skin Whitening Agents. Int. J. Mol. Sci. 2009, 10, 5326–5349. [Google Scholar] [CrossRef]
- Xin, L.-T.; Yue, S.-J.; Fan, Y.-C.; Wu, J.-S.; Yan, D.; Guan, H.-S.; Wang, C.-Y. Cudrania Tricuspidata: An Updated Review on Ethnomedicine, Phytochemistry and Pharmacology. RSC Adv. 2017, 7, 31807–31832. [Google Scholar] [CrossRef] [Green Version]
- Wink, M. Evolution of Secondary Metabolites from an Ecological and Molecular Phylogenetic Perspective. Phytochemistry 2003, 64, 3–19. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, K.; Mizokami, A.; Lin, W.-J.; Lai, K.-P.; Chang, C. Androgen Receptor Roles in the Development of Benign Prostate Hyperplasia. Am. J. Pathol. 2013, 182, 1942–1949. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E. Androgen Receptor: Structure, Role in Prostate Cancer and Drug Discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liss, M.A.; Thompson, I.M. Prostate Cancer Prevention with 5-Alpha Reductase Inhibitors: Concepts and Controversies. Curr. Opin. Urol. 2018, 28, 42–45. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Nodes | Number of Edges | Density 1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Plant (Np) | Chemical (Nc) | Target (Nt) | Total (N) | Plant-Chemical | Chemical-Target | Total (E) | ||
| Raw Data | 5500 | 10,056 | 1224 | 16,780 | 58,068 | 100,290 | 158,358 | 0.00234 |
| without Non-plants | 3614 | 10,056 | 1224 | 14,894 | 54,585 | 100,290 | 154,875 | 0.00318 |
| without Non-plants or Duplicates | 2886 | 10,056 | 1224 | 14,166 | 34,549 | 100,290 | 134,839 | 0.00326 |
| Only targets with the probability of above 0.9 | 2886 | 10,056 | 441 | 13,383 | 34,549 | 73,112 | 107,661 | 0.00322 |
| Pre-processed Data | 1138 | 10,043 | 441 | 11,622 | 34,549 | 73,112 | 107,661 | 0.00679 |
| Chemical Name | CAS Number | Chemicals Classification | ||||
|---|---|---|---|---|---|---|
| Kingdom | Superclass | Class | Subclass | Direct Parent | ||
| Kusunol | 20489-45-6 | Organic compounds | Lipids and lipid-like molecules | Prenol lipids | Sesquiterpenoids | Eremophilane, 8,9-secoeremophilane and furoeremophilane sesquiterpenoids |
| Stigmasta-4,6-dien-3-one | 29374-98-9 | Organic compounds | Lipids and lipid-like molecules | Steroids and steroid derivatives | Stigmastanes and derivatives | Stigmastanes and derivatives |
| Stigmastan-3-one (5-alpha) | 102734-69-0 | Organic compounds | Lipids and lipid-like molecules | Steroids and steroid derivatives | Stigmastanes and derivatives | Stigmastanes and derivatives |
| Dehydroabietinal | 13601-88-2 | Organic compounds | Lipids and lipid-like molecules | Prenol lipids | Diterpenoids | Diterpenoids |
| (-)-alpha-Copaene | 3856-25-5 | Organic compounds | Lipids and lipid-like molecules | Prenol lipids | Sesquiterpenoids | Sesquiterpenoids |
| (-)-Ylangene | 14912-44-8 | Organic compounds | Lipids and lipid-like molecules | Prenol lipids | Sesquiterpenoids | Sesquiterpenoids |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, N.; Yoo, H.; Yang, H. Cluster Analysis of Medicinal Plants and Targets Based on Multipartite Network. Biomolecules 2021, 11, 546. https://doi.org/10.3390/biom11040546
Lee N, Yoo H, Yang H. Cluster Analysis of Medicinal Plants and Targets Based on Multipartite Network. Biomolecules. 2021; 11(4):546. https://doi.org/10.3390/biom11040546
Chicago/Turabian StyleLee, Namgil, Hojin Yoo, and Heejung Yang. 2021. "Cluster Analysis of Medicinal Plants and Targets Based on Multipartite Network" Biomolecules 11, no. 4: 546. https://doi.org/10.3390/biom11040546
APA StyleLee, N., Yoo, H., & Yang, H. (2021). Cluster Analysis of Medicinal Plants and Targets Based on Multipartite Network. Biomolecules, 11(4), 546. https://doi.org/10.3390/biom11040546