
A Comprehensive Review of Cholinesterase Modeling and Simulation



Abstract
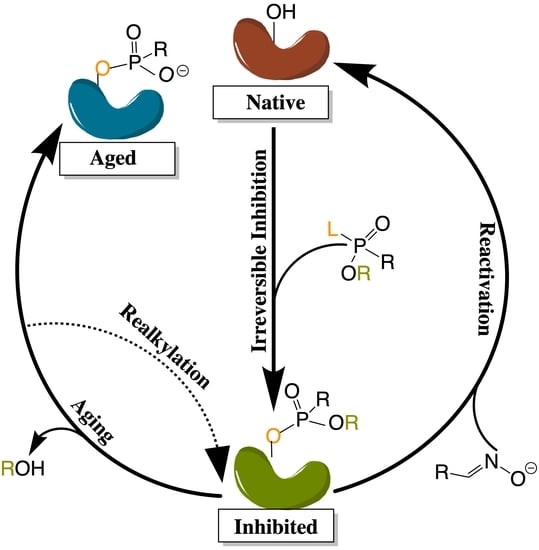
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Structure and Dynamics
2.1. Important Binding Sites
2.1.1. Peripheral Aromatic Site
2.1.2. Acyl and Choline Binding Sites
2.1.3. Catalytic Active Site
2.1.4. Oxyanion Hole
2.1.5. Ω-loop (Omega Loop)
3. Catalysis
4. Inhibition
4.1. Pharmaceuticals
4.1.1. Tacrine and Derivatives
4.1.2. Galantamine and Derivatives
4.1.3. Donepezil and Derivatives
4.1.4. Rivastigmine and Derivatives
4.1.5. Quinazoline and Derivatives
4.1.6. Coumarin
4.1.7. Other Pharmaceuticals
4.2. Narcotics
4.3. Organophosphates
4.3.1. Reversible Inhibition
4.3.2. Irreversible Inhibition, Activation, and Reactivation
4.4. Other Organic Moieties
4.4.1. Hydrocarbons
4.4.2. Nitrogenous Compounds
Amines
Amides, Imides, Imines, and Carbamates
Nitrogenous Heterocyclic Rings and Derivatives
4.4.3. Organosulfates
4.5. Proteins, Nucleic Acids, and Salts
4.5.1. Protein and RNA Binding
4.5.2. Nucleobase Derivatives
4.5.3. Ion and Salt Binding
5. Virtual Screening
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, P.; Camp, S.; Radić, Z. Acetylcholinesterase. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, UK, 2009. [Google Scholar] [CrossRef]
- John, H.; Balszuweit, F.; Kehe, K.; Worek, F.; Thiermann, H. Chapter 50—Toxicokinetics of Chemical Warfare Agents: Nerve Agents and Vesicants. In Handbook of Toxicology of Chemical Warfare Agents; Gupta, R.C., Ed.; Academic Press: San Diego, CA, USA, 2009. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S.W. Why has butyrylcholinesterase been retained? Structural and functional diversification in a duplicated gene. Neurochem. Int. 2012, 61, 783–797. [Google Scholar] [CrossRef]
- Ortiz, J.E.; Pigni, N.B.; Andujar, S.A.; Roitman, G.; Suvire, F.D.; Enriz, R.D.; Tapia, A.; Bastida, J.; Feresin, G.E. Alkaloids from Hippeastrum argentinum and Their Cholinesterase-Inhibitory Activities: An in Vitro and in Silico Study. J. Nat. Prod. 2016, 79, 1241–1248. [Google Scholar] [CrossRef]
- Masson, P.; Lushchekina, S.; Schopfer, L.M.; Lockridge, O. Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: Kinetic and molecular dynamics studies. Biochem. J. 2013, 454, 387–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Barron, M.G. A mechanism-based 3D-QSAR approach for classification and prediction of acetylcholinesterase inhibitory potency of organophosphate and carbamate analogs. J. Comput. Aided Mol. Des. 2016, 30, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Kharlamova, A.D.; Lushchekina, S.V.; Petrov, K.A.; Kots, E.D.; Nachon, F.; Villard-Wandhammer, M.; Zueva, I.V.; Krejci, E.; Reznik, V.S.; Zobov, V.V.; et al. Slow-binding inhibition of acetylcholinesterase by an alkylammonium derivative of 6-methyluracil: Mechanism and possible advantages for myasthenia gravis treatment. Biochem. J. 2016, 473, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.E.; Botti, S.A.; Lifson, S.; Silman, I.; Sussman, J.L. External and internal electrostatic potentials of cholinesterase models. J. Mol. Graph. Model. 1997, 15, 318–327. [Google Scholar] [CrossRef]
- Ochoa, R.; Rodriguez, C.A.; Zuluaga, A.F. Perspectives for the structure-based design of acetylcholinesterase reactivators. J. Mol. Graph. Model. 2016, 68, 176–183. [Google Scholar] [CrossRef]
- Saxena, A.; Redman, A.M.G.; Jiang, X.; Lockridge, O.; Doctor, B.P. Differences in Active Site Gorge Dimensions of Cholinesterases Revealed by Binding of Inhibitors to Human Butyrylcholinesterase. Biochemistry 1997, 36, 14642–14651. [Google Scholar] [CrossRef]
- Ul-Haq, Z.; Khan, W.; Kalsoom, S.; Ansari, F.L. In silico modeling of the specific inhibitory potential of thiophene-2,3-dihydro-1,5-benzothiazepine against BChE in the formation of beta-amyloid plaques associated with Alzheimer’s disease. Theor. Biol. Med. Model. 2010, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Sussman, J.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal Structure of Human Butyrylcholinesterase and of Its Complexes with Substrate and Products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Q.G.; Young, A.; Callam, C.S.; McElroy, C.A.; Ekici, O.D.; Yoder, R.J.; Hadad, C.M. Efforts toward treatments against aging of organophosphorus-inhibited acetylcholinesterase. Ann. N. Y. Acad. Sci. 2016, 1374, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Q.G.; Franjesevic, A.J.; Corrigan, T.S.; Coldren, W.H.; Dicken, R.; Sillart, S.; DeYong, A.; Yoshino, N.; Smith, J.; Fabry, S.; et al. Demonstration of In Vitro Resurrection of Aged Acetylcholinesterase after Exposure to Organophosphorus Chemical Nerve Agents. J. Med. Chem. 2018, 61, 7034–7042. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.M. Resurrection Biology: Aged Acetylcholinesterase Brought Back to Life. J. Med. Chem. 2018, 61, 7032–7033. [Google Scholar] [CrossRef] [Green Version]
- Suárez, D.; Field, M.J. Molecular dynamics simulations of human butyrylcholinesterase. Proteins Struct. Funct. Bioinform. 2005, 59, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-X.; Wlodek, S.T.; McCammon, J.A. Conformation gating as a mechanism for enzyme specificity. Proc. Natl. Acad. Sci. USA 1998, 95, 9280–9283. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Tai, K.; Henchman, R.H.; McCammon, J.A. Molecular Dynamics of Acetylcholinesterase. Acc. Chem. Res. 2002, 35, 332–340. [Google Scholar] [CrossRef]
- Cheng, S.M.; Song, W.L.; Yuan, X.J.; Xu, Y.C. Gorge Motions of Acetylcholinesterase Revealed by Microsecond Molecular Dynamics Simulations. Sci. Rep. 2017, 7, 3219. [Google Scholar] [CrossRef]
- Xu, Y.; Colletier, J.-P.; Weik, M.; Jiang, H.; Moult, J.; Silman, I.; Sussman, J.L. Flexibility of Aromatic Residues in the Active-Site Gorge of Acetylcholinesterase: X-ray versus Molecular Dynamics. Biophys. J. 2008, 95, 2500–2511. [Google Scholar] [CrossRef] [Green Version]
- Chinnadurai, R.K.; Saravanaraman, P.; Boopathy, R. Understanding the molecular mechanism of aryl acylamidase activity of acetylcholinesterase—An in silico study. Arch. Biochem. Biophys. 2015, 580, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Axelsen, P.H.; Harel, M.; Silman, I.; Sussman, J.L. Structure and dynamics of the active site gorge of acetylcholinesterase: Synergistic use of molecular dynamics simulation and X-ray crystallography. Protein Sci. 1994, 3, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henchman, R.H.; McCammon, J.A. Structural and dynamic properties of water around acetylcholinesterase. Protein Sci. 2002, 11, 2080–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koellner, G.; Kryger, G.; Millard, C.B.; Silman, I.; Sussman, J.L.; Steiner, T. Active-site gorge and buried water molecules in crystal structures of acetylcholinesterase from Torpedo californica. J. Mol. Biol. 2000, 296, 713–735. [Google Scholar] [CrossRef] [Green Version]
- Leung, M.R.; van Bezouwen, L.S.; Schopfer, L.M.; Sussman, J.L.; Silman, I.; Lockridge, O.; Zeev-Ben-Mordehaia, T. Cryo-EM structure of the native butyrylcholinesterase tetramer reveals a dimer of dimers stabilized by a superhelical assembly. Proc. Natl. Acad. Sci. USA 2018, 115, 13270–13275. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Pan, Y.; Muzyka, J.L.; Zhan, C.-G. Active Site Gating and Substrate Specificity of Butyrylcholinesterase and Acetylcholinesterase: Insights from Molecular Dynamics Simulations. J. Phys. Chem. B 2011, 115, 8797–8805. [Google Scholar] [CrossRef] [Green Version]
- Gorfe, A.A.; Lu, B.Z.; Yu, Z.Y.; McCammon, J.A. Enzymatic Activity versus Structural Dynamics: The Case of Acetylcholinesterase Tetramer. Biophys. J. 2009, 97, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Muzyka, J.L.; Zhan, C.-G. Model of Human Butyrylcholinesterase Tetramer by Homology Modeling and Dynamics Simulation. J. Phys. Chem. B 2009, 113, 6543–6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Zheng, F.; Zhan, C.-G. A model of glycosylated human butyrylcholinesterase. Mol. Biosyst. 2014, 10, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Bennion, B.J.; Essiz, S.G.; Lau, E.Y.; Fattebert, J.-L.; Emigh, A.; Lightstone, F.C. A wrench in the works of human acetylcholinesterase: Soman induced conformational changes revealed by molecular dynamics simulations. PLoS ONE 2015, 10, e0121092. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.; Martinez, N.; Trovaslet, M.; Scannapieco, K.; Koza, M.M.; Masson, P.; Nachon, F. Dynamics of human acetylcholinesterase bound to non-covalent and covalent inhibitors shedding light on changes to the water network structure. Phys. Chem. Chem. Phys. 2016, 18, 12992–13001. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarado, W.; Bremer, P.L.; Choy, A.; Dinh, H.N.; Eung, A.; Gonzalez, J.; Ly, P.; Tran, T.; Nakayama, K.; Schwans, J.P.; et al. Understanding the enzyme-ligand complex: Insights from all-atom simulations of butyrylcholinesterase inhibition. J. Biomol. Struct. Dyn. 2019, 38, 1028–1041. [Google Scholar] [CrossRef]
- Bourne, Y.; Taylor, P.; Bougis, P.E.; Marchot, P. Crystal structure of mouse acetylcholinesterase—A peripheral site-occluding loop in a tetrameric assembly. J. Biol. Chem. 1999, 274, 2963–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campiani, G.; Fattorusso, C.; Butini, S.; Gaeta, A.; Agnusdei, M.; Gemma, S.; Persico, M.; Catalanotti, B.; Savini, L.; Nacci, V.; et al. Development of molecular probes for the identification of extra interaction sites in the mid-gorge and peripheral sites of butyrylcholinesterase (BuChE). Rational design of novel, selective, and highly potent BuChE inhibitors. J. Med. Chem. 2005, 48, 1919–1929. [Google Scholar] [CrossRef]
- Khan, M.T.H. Molecular interactions of cholinesterases inhibitors using in silico methods: Current status and future prospects. New Biotechnol. 2009, 25, 331–346. [Google Scholar] [CrossRef]
- Roca, C.; Requena, C.; Sebastian-Perez, V.; Malhotra, S.; Radoux, C.; Perez, C.; Martinez, A.; Antonio Paez, J.; Blundell, T.L.; Campillo, N.E. Identification of new allosteric sites and modulators of AChE through computational and experimental tools. J. Enzym. Inhib. Med. Chem. 2018, 33, 1034–1047. [Google Scholar] [CrossRef] [Green Version]
- Branduardi, D.; Gervasio, F.L.; Cavalli, A.; Recanatini, M.; Parrinello, M. The role of the peripheral anionic site and cation-pi interactions in the ligand penetration of the human AChE gorge. J. Am. Chem. Soc. 2005, 127, 9147–9155. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S. The peripheral anionic site of acetylcholinesterase: Structure, functions and potential role in rational drug design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Dighe, S.N.; Deora, G.S.; De la Mora, E.; Nachon, F.; Chan, S.; Parat, M.O.; Brazzolotto, X.; Ross, B.P. Discovery and Structure-Activity Relationships of a Highly Selective Butyrylcholinesterase Inhibitor by Structure-Based Virtual Screening. J. Med. Chem. 2016, 59, 7683–7689. [Google Scholar] [CrossRef] [Green Version]
- Kwong, H.C.; Chidan Kumar, C.S.; Mah, S.H.; Mah, Y.L.; Chia, T.S.; Quah, C.K.; Lim, G.K.; Chandraju, S. Crystal Correlation Of Heterocyclic Imidazo[1,2-a]pyridine Analogues and Their Anticholinesterase Potential Evaluation. Sci. Rep. 2019, 9, 926. [Google Scholar] [CrossRef]
- Kumar, J.; Gill, A.; Shaikh, M.; Singh, A.; Shandilya, A.; Jameel, E.; Sharma, N.; Mrinal, N.; Hoda, N.; Jayaram, B. Pyrimidine-Triazolopyrimidine and Pyrimidine-Pyridine Hybrids as Potential Acetylcholinesterase Inhibitors for Alzheimer’s Disease. ChemistrySelect 2018, 3, 736–747. [Google Scholar] [CrossRef]
- Bencsura, A.; Enyedy, I.Y.; Kovach, I.M. Probing the active site of acetylcholinesterase by molecular dynamics of its phosphonate ester adducts. J. Am. Chem. Soc. 1996, 118, 8531–8541. [Google Scholar] [CrossRef]
- Viragh, C.; Harris, T.K.; Reddy, P.M.; Massiah, M.A.; Mildvan, A.S.; Kovach, I.M. NMR Evidence for a Short, Strong Hydrogen Bond at the Active Site of a Cholinesterase. Biochemistry 2000, 39, 16200–16205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Kua, J.; McCammon, J.A. Role of the catalytic triad and oxyanion hole in acetylcholinesterase catalysis: An ab initio QM/MM study. J. Am. Chem. Soc. 2002, 124, 10572–10577. [Google Scholar] [CrossRef] [PubMed]
- Kua, J.; Zhang, Y.; McCammon, J.A. Studying Enzyme Binding Specificity in Acetylcholinesterase Using a Combined Molecular Dynamics and Multiple Docking Approach. J. Am. Chem. Soc. 2002, 124, 8260–8267. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Straatsma, T.P.; McCammon, J.A.; Ripoli, D.R.; Faerman, C.H.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science 1994, 263, 1276–1278. [Google Scholar] [CrossRef]
- Sanson, B.; Colletier, J.-P.; Xu, Y.; Lang, P.T.; Jiang, H.; Silman, I.; Sussman, J.L.; Weik, M. Backdoor opening mechanism in acetylcholinesterase based on X-ray crystallography and molecular dynamics simulations. Protein Sci. 2011, 20, 1114–1118. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.; Barak, D.; Ordentlich, A.; Kronman, C.; Velan, B.; Shafferman, A. Is aromaticity essential for trapping the catalytic histidine 447 in human acetylcholinesterase? Biochemistry 2004, 43, 3129–3136. [Google Scholar] [CrossRef] [PubMed]
- Barak, D.; Kaplan, D.; Ordentlich, A.; Ariel, N.; Velan, B.; Shafferman, A. The aromatic “trapping” of the catalytic histidine is essential for efficient catalysis in acetylcholinesterase. Biochemistry 2002, 41, 8245–8252. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Colletier, J.-P.; Weik, M.; Qin, G.; Jiang, H.; Silman, I.; Sussman, J.L. Long route or shortcut? A molecular dynamics study of traffic of thiocholine within the active-site gorge of acetylcholinesterase. Biophys. J. 2010, 99, 4003–4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colletier, J.; Royant, A.; Specht, A.; Sanson, B.; Nachon, F.; Masson, P.; Zaccai, G.; Sussman, J.; Goeldner, M.; Silman, I.; et al. Use of a ‘caged’ analogue to study the traffic of choline within acetylcholinesterase by kinetic crystallography. Acta Crystallogr. D 2007, 63, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.-P.; Bourgeois, D.; Sanson, B.; Fournier, D.; Sussman, J.L.; Silman, I.; Weik, M. Shoot-and-Trap: Use of specific X-ray damage to study structural protein dynamics by temperature-controlled cryo-crystallography. Proc. Natl. Acad. Sci. USA 2008, 105, 11742–11747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delacour, H.; Lushchekina, S.; Mabboux, I.; Ceppa, F.; Masson, P.; Schopfer, L.M.; Lockridge, O. Characterization of a novel butyrylcholinesterase point mutation (p.Ala34Val), “silent” with mivacurium. Biochem. Pharmacol. 2014, 92, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Delacour, H.; Lushchekina, S.; Mabboux, I.; Bousquet, A.; Ceppa, F.; Schopfer, L.M.; Lockridge, O.; Masson, P. Characterization of a Novel BCHE “Silent’’ Allele: Point Mutation (p. Val204Asp) Causes Loss of Activity and Prolonged Apnea with Suxamethonium. PLoS ONE 2014, 9, e101552. [Google Scholar] [CrossRef] [Green Version]
- Lushchekina, S.V.; Nemukhin, A.V.; Varfolomeev, S.D.; Masson, P. Molecular Modeling Evidence for His438 Flip in the Mechanism of Butyrylcholinesterase Hysteretic Behavior. J. Mol. Neurosci. 2014, 52, 434–445. [Google Scholar] [CrossRef]
- Lushchekina, S.; Nemukhin, A.; Varfolomeev, S.; Masson, P. Understanding the non-catalytic behavior of human butyrylcholinesterase silent variants: Comparison of wild-type enzyme, catalytically active Ala328Cys mutant, and silent Ala328Asp variant. Chem. Biol. Interact. 2016, 259, 223–232. [Google Scholar] [CrossRef]
- Grigorenko, B.L.; Novichkova, D.A.; Lushchekina, S.V.; Zueva, I.V.; Schopfer, L.M.; Nemukhin, A.V.; Varfolomeev, S.D.; Lockridge, O.; Masson, P. Computer-designed active human butyrylcholinesterase double mutant with a new catalytic triad. Chem. Biol. Interact. 2019, 306, 138–146. [Google Scholar] [CrossRef]
- Zheng, F.; Yang, W.C.; Xue, L.; Hou, S.R.; Liu, J.J.; Zhan, C.G. Design of High-Activity Mutants of Human Butyrylcholinesterase against (-)-Cocaine: Structural and Energetic Factors Affecting the Catalytic Efficiency. Biochemistry 2010, 49, 9113–9119. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Zhan, C.-G. Modeling Effects of Oxyanion Hole on the Ester Hydrolysis Catalyzed by Human Cholinesterases. J. Phys. Chem. B 2005, 109, 23070–23076. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.X.; Tai, K.; McCammon, J.A.; Taylor, P.; Johnson, D.A. Nanosecond dynamics of the mouse acetylcholinesterase Cys(69)-Cys(96) omega loop. J. Biol. Chem. 2003, 278, 30905–30911. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Boyd, A.E.; Radic, Z.R.; Taylor, P. Reversibly Bound and Covalently Attached Ligands Induce Conformational Changes in the Omega Loop, Cys69–Cys96, of Mouse Acetylcholinesterase*. J. Biol. Chem. 2001, 276, 42196–42204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, P.; Legrand, P.; Bartels, C.F.; Froment, M.T.; Schopfer, L.M.; Lockridge, O. Role of aspartate 70 and tryptophan 82 in binding of succinyldithiocholine to human butyrylcholinesterase. Biochemistry 1997, 36, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Kriz, Z.; Kuca, K.; Jun, D.; Koca, J. Influence of the Acetylcholinesterase Active Site Protonation on Omega Loop and Active Site Dynamics. J. Biomol. Struct. Dyn. 2010, 28, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Rydzewski, J.; Jakubowski, R.; Nowak, W.; Grubmuller, H. Kinetics of Huperzine A Dissociation from Acetylcholinesterase via Multiple Unbinding Pathways. J. Chem. Theory Comput. 2018, 14, 2843–2851. [Google Scholar] [CrossRef] [Green Version]
- Bourne, Y.; Renault, L.; Marchot, P. Crystal Structure of Snake Venom Acetylcholinesterase in Complex with Inhibitory Antibody Fragment Fab410 Bound at the Peripheral Site evidence for open and closed states of a back door channel. J. Biol. Chem. 2015, 290, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Rosenberry, T.L.; Silman, I.; Sussman, J.L. A Second Look at the Crystal Structures of Drosophila melanogaster Acetylcholinesterase in Complex with Tacrine Derivatives Provides Insights Concerning Catalytic Intermediates and the Design of Specific Insecticides. Molecules 2020, 25, 1198. [Google Scholar] [CrossRef] [Green Version]
- Nachon, F.; Stojan, J.; Fournier, D. Insights into substrate and product traffic in the Drosophila melanogaster acetylcholinesterase active site gorge by enlarging a back channel. FEBS J. 2008, 275, 2659–2664. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Zhang, Y. Catalytic reaction mechanism of acetylcholinesterase determined by born-oppenheimer ab initio QM/MM molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 8817–8825. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Fang, L.; Liu, J.J.; Zhan, C.G. Reaction Pathway and Free Energy Profile for Butyrylcholinesterase-Catalyzed Hydrolysis of Acetylcholine. J. Phys. Chem. B 2011, 115, 1315–1322. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Fang, L.; Liu, J.J.; Zhan, C.G. Reaction Pathway and Free Energy Profiles for Butyrylcholinesterase-Catalyzed Hydrolysis of Acetylthiocholine. Biochemistry 2012, 51, 1297–1305. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; Han, K.; Zhan, C.-G. Fundamental Reaction Pathway and Free Energy Profile for Butyrylcholinesterase-Catalyzed Hydrolysis of Heroin. Biochemistry 2013, 52, 6467–6479. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Yuan, Y.; Zheng, F.; Zhan, C.-G. Unexpected Reaction Pathway for butyrylcholinesterase-catalyzed inactivation of “hunger hormone” ghrelin. Sci. Rep. 2016, 6, 22322. [Google Scholar] [CrossRef]
- Suarez, D.; Diaz, N.; Fontecilla-Camps, J.; Field, M.J. A computational study of the deacylation mechanism of human butyrylcholinesterase. Biochemistry 2006, 45, 7529–7543. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Pan, Y.; Zheng, F.; Zhan, C.-G. Reaction Pathway and Free Energy Profile for Prechemical Reaction Step of Human Butyrylcholinesterase-Catalyzed Hydrolysis of (-)-Cocaine by Combined Targeted Molecular Dynamics and Potential of Mean Force Simulations. J. Phys. Chem. B 2010, 114, 13545–13554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, C.G.; Gao, D.Q. Catalytic mechanism and energy barriers for butyrylcholinesterase-catalyzed hydrolysis of cocaine. Biophys. J. 2005, 89, 3863–3872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Gao, D.; Yang, W.; Cho, H.; Yang, G.; Tai, H.-H.; Zhan, C.-G. Computational redesign of human butyrylcholinesterase for anticocaine medication. Proc. Natl. Acad. Sci. USA 2005, 102, 16656–16661. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Pan, Y.; Zheng, F.; Cho, H.; Tai, H.-H.; Zhan, C.-G. Free-Energy Perturbation Simulation on Transition States and Redesign of Butyrylcholinesterase. Biophys. J. 2009, 96, 1931–1938. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.M.; Gao, D.Q.; Yang, W.C.; Cho, H.; Zhan, C.G. Free energy perturbation (FEP) simulation on the transition states of cocaine hydrolysis catalyzed by human butyrylcholinesterase and its mutants. J. Am. Chem. Soc. 2007, 129, 13537–13543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Zheng, F.; Zhan, C.-G. Human butyrylcholinesterase-cocaine binding pathway and free energy profiles by molecular dynamics and potential of mean force simulations. J. Phys. Chem. B 2011, 115, 11254–11260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharmacal Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer Disease. Dis. Mon. 2010, 56, 484–546. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.D.; Faraj, F.L.; Lee, V.S.; Othman, R.; Buckle, M.J.C. 2-Aryl-3-(arylideneamino)-1,2-dihydroquinazoline-4(3H)-ones as inhibitors of cholinesterases and self-induced beta-amyloid (Ab) aggregation: Biological evaluations and mechanistic insights from molecular dynamics simulations. RSC Adv. 2018, 8, 7818–7831. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Hu, J.; Wang, L.; Zhong, G.; Pan, J.; Wu, Z.; Hui, A. Combined 3D-QSAR, molecular docking, and molecular dynamics study of tacrine derivatives as potential acetylcholinesterase (AChE) inhibitors of Alzheimer’s disease. J. Mol. Modeling 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Maalej, E.; Chabchoub, F.; Samadi, A.; de los Rios, C.; Perona, A.; Morreale, A.; Marco-Contelles, J. Synthesis, biological assessment and molecular modeling of 14-aryl-10,11,12,14-tetrahydro-9H-benzo 5,6 chromeno 2,3-b quinolin-13-amines. Bioorg. Med. Chem. Lett. 2011, 21, 2384–2388. [Google Scholar] [CrossRef]
- Thiratmatrakul, S.; Yenjai, C.; Waiwut, P.; Vajragupta, O.; Reubroycharoen, P.; Tohda, M.; Boonyarat, C. Synthesis, biological evaluation and molecular modeling study of novel tacrine–carbazole hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 75, 21–30. [Google Scholar] [CrossRef]
- Manetsch, R.; Krasiński, A.; Radić, Z.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In Situ Click Chemistry: Enzyme Inhibitors Made to Their Own Specifications. J. Am. Chem. Soc. 2004, 126, 12809–12818. [Google Scholar] [CrossRef]
- Zhu, X.-L.; Yu, N.-X.; Hao, G.-F.; Yang, W.-C.; Yang, G.-F. Structural basis of femtomolar inhibitors for acetylcholinesterase subtype selectivity: Insights from computational simulations. J. Mol. Graph. Model. 2013, 41, 55–60. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Rudakova, E.V.; Stupina, T.S.; Terentiev, A.A.; Serkov, I.V.; Proshin, A.N.; Radchenko, E.V.; et al. Conjugates of tacrine and 1,2,4-thiadiazole derivatives as new potential multifunctional agents for Alzheimer’s disease treatment: Synthesis, quantum-chemical characterization, molecular docking, and biological evaluation. Bioorg. Chem. 2020, 94, 103387. [Google Scholar] [CrossRef]
- Chen, X.; Wehle, S.; Kuzmanovic, N.; Merget, B.; Holzgrabe, U.; Konig, B.; Sotriffer, C.A.; Decker, M. Acetylcholinesterase Inhibitors with Photoswitchable Inhibition of beta-Amyloid Aggregation. ACS Chem. Neurosci. 2014, 5, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.Q.; Zhu, K.K.; Zhang, J.; Song, J.L.; Muehlmann, L.A.; Jiang, C.S.; Liu, C.L.; Zhang, H. Molecular-docking-guided design and synthesis of new IAA-tacrine hybrids as multifunctional AChE/BChE inhibitors. Bioorg. Chem. 2019, 83, 277–288. [Google Scholar] [CrossRef]
- Nascimento, E.C.M.; Oliva, M.; Swiderek, K.; Martins, J.B.L.; Andres, J. Binding Analysis of Some Classical Acetylcholinesterase Inhibitors: Insights for a Rational Design Using Free Energy Perturbation Method Calculations with QM/MM MD Simulations. J. Chem. Inf. Modeling 2017, 57, 958–976. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Yao, Y.; Fang, L.; Liu, J.J. Unexpected protonation state of Glu197 discovered from simulations of tacrine in butyrylcholinesterase. Phys. Chem. Chem. Phys. 2018, 20, 14938–14946. [Google Scholar] [CrossRef]
- Galdeano, C.; Viayna, E.; Sola, I.; Formosa, X.; Camps, P.; Badia, A.; Clos, M.V.; Relat, J.; Ratia, M.; Bartolini, M.; et al. Huprine–Tacrine Heterodimers as Anti-Amyloidogenic Compounds of Potential Interest against Alzheimer’s and Prion Diseases. J. Med. Chem. 2012, 55, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Eslami, M.; Hashemianzadeh, S.M.; Moghaddam, K.G.; Khorsandi-Lagol, A.; Seyed Sajadi, S.A. Computational evidence to design an appropriate candidate for the treatment of Alzheimer’s disease through replacement of the heptamethylene linker of bis(7)tacrine with S-allylcysteine. RSC Adv. 2015, 5, 66840–66851. [Google Scholar] [CrossRef]
- Eslami, M.; Hashemianzadeh, S.M.; Bagherzadeh, K.; Sajadi, S.A.S. Molecular perception of interactions between bis(7)tacrine and cystamine-tacrine dimer with cholinesterases as the promising proposed agents for the treatment of Alzheimer’s disease. J. Biomol. Struct. Dyn. 2015, 34, 855–869. [Google Scholar] [CrossRef]
- Habibpour, R.; Eslami, M.; Amani, P.; Novir, S.B. Tacrine-flavonoid quercetin hybride as a MTDL ligand against alzheimer’s disease with metal chelating and AChE, BChE, AChE-induced Aβ aggregation inhibition properties: A computational study. Phys. Chem. Res. 2019, 7, 561–579. [Google Scholar]
- Brito, M.d.F.d.B.; Ferreira, J.V.; de Souza, L.R.; Gemaque, L.R.P.; Sousa, K.P.A.; dos Santos, C.F.; Braga, F.S.; Pernomian, L.; da Silva, C.H.T.P.; Santos, C.B.R.; et al. Computational Molecular Modeling of Compounds from Amaryllidaceae Family as Potential Acetylcholinesterase Inhibitors. Curr. Bioact. Compd. 2017, 13, 121–129. [Google Scholar] [CrossRef]
- Gulcan, H.O.; Orhan, I.E.; Sener, B. Chemical and Molecular Aspects on Interactions of Galanthamine and Its Derivatives with Cholinesterases. Curr. Pharm. Biotechnol. 2015, 16, 252–258. [Google Scholar] [CrossRef]
- Ali, M.R.; Sadoqi, M.; Møller, S.G.; Boutajangout, A.; Mezei, M. Assessing the binding of cholinesterase inhibitors by docking and molecular dynamics studies. J. Mol. Graph. Model. 2017, 76, 36–42. [Google Scholar] [CrossRef]
- Silva, M.A.; Kiametis, A.S.; Treptow, W. Donepezil Inhibits Acetylcholinesterase via Multiple Binding Modes at Room Temperature. J. Chem. Inf. Modeling 2020, 60, 3463–3471. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, O.M.; Esteban, G.; Chioua, M.; Nikolic, K.; Agbaba, D.; Moraleda, I.; Iriepa, I.; Soriano, E.; Samadi, A.; Unzeta, M.; et al. Multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease: Design, synthesis, biochemical evaluation, ADMET, molecular modeling, and QSAR analysis of novel donepezil-pyridyl hybrids. Drug Des. Dev. Ther. 2014, 8, 1893–1910. [Google Scholar]
- Valasani, K.R.; Chaney, M.O.; Day, V.W.; Yan, S.S. Acetylcholinesterase Inhibitors: Structure Based Design, Synthesis, Pharmacophore Modeling, and Virtual Screening. J. Chem. Inf. Modeling 2013, 53, 2033–2046. [Google Scholar] [CrossRef]
- Silva, D.; Chioua, M.; Samadi, A.; Agostinho, P.; Garcao, P.; Lajarin-Cuesta, R.; de los Rios, C.; Iriepa, I.; Moraleda, I.; Gonzalez-Lafuente, L.; et al. Synthesis, Pharmacological Assessment, and Molecular Modeling of Acetylcholinesterase/Butyrylcholinesterase Inhibitors: Effect against Amyloid-beta-Induced Neurotoxicity. ACS Chem. Neurosci. 2013, 4, 547–565. [Google Scholar] [CrossRef] [Green Version]
- Al-Rashid, Z.F.; Hsung, R.P. A computational view on the significance of E-ring in binding of (+)-arisugacin A to acetylcholinesterase. Bioorg. Med. Chem. Lett. 2015, 25, 4848–4853. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Ali, M.T.; Shawan, M.M.A.K.; Sarwar, M.G.; Khan, M.A.K.; Halim, M.A. Halogen-directed drug design for Alzheimer’s disease: A combined density functional and molecular docking study. SpringerPlus 2016, 5, 1346. [Google Scholar] [CrossRef] [Green Version]
- Alpan, A.S.; Parlar, S.; Carlino, L.; Tarikogullari, A.H.; Alptüzün, V.; Güneş, H.S. Synthesis, biological activity and molecular modeling studies on 1H-benzimidazole derivatives as acetylcholinesterase inhibitors. Bioorg. Med. Chem. 2013, 21, 4928–4937. [Google Scholar] [CrossRef]
- Ghosh, S.; Jana, K.; Ganguly, B. Revealing the mechanistic pathway of cholinergic inhibition of Alzheimer’s disease by donepezil: A metadynamics simulation study. Phys. Chem. Chem. Phys. 2019, 21, 13578–13589. [Google Scholar] [CrossRef]
- Bolea, I.; Juárez-Jiménez, J.; de los Ríos, C.; Chioua, M.; Pouplana, R.; Luque, F.J.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, Biological Evaluation, and Molecular Modeling of Donepezil and N-[(5-(Benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine Hybrids as New Multipotent Cholinesterase/Monoamine Oxidase Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2011, 54, 8251–8270. [Google Scholar] [PubMed]
- Yekta, R.; Sadeghi, L.; Dehghan, G. The inefficacy of donepezil on glycated-AChE inhibition: Binding affinity, complex stability and mechanism. Int. J. Biol. Macromol. 2020, 160, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Bartolini, M.; Cavalli, A.; Andrisano, V.; Rosini, M.; Minarini, A.; Melchiorre, C. Design, Synthesis, and Biological Evaluation of Conformationally Restricted Rivastigmine Analogues. J. Med. Chem. 2004, 47, 5945–5952. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Tian, Y.G.; Shang, J.L.; Sun, X.O.; Chen, H.Z.; Wang, H.; Tan, W. Design, synthesis, biological evaluation, and molecular modeling studies of chalcone-rivastigmine hybrids as cholinesterase inhibitors. Bioorg. Med. Chem. 2017, 25, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Hekal, M.H.; Abu El-Azm, F.S.M. New potential antitumor quinazolinones derived from dynamic 2-undecyl benzoxazinone: Synthesis and cytotoxic evaluation. Synth. Commun. 2018, 48, 2391–2402. [Google Scholar] [CrossRef]
- Li, Z.; Wang, B.; Hou, J.Q.; Huang, S.L.; Ou, T.M.; Tan, J.H.; An, L.K.; Li, D.; Gu, L.Q.; Huang, Z.S. 2-(2-indolyl-)-4(H)-quinazolines derivates as new inhibitors of AChE: Design, synthesis, biological evaluation and molecular modelling. J. Enzym. Inhib. Med. Chem. 2013, 28, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Abdul Hameed, M.D.M.; Liu, J.; Pan, Y.; Fang, L.; Silva-Rivera, C.; Zhan, C.-G. Microscopic binding of butyrylcholinesterase with quinazolinimine derivatives and the structure-activity correlation. Theor. Chem. Acc. 2011, 130, 69–82. [Google Scholar] [CrossRef]
- Chen, X.; Tikhonova, I.G.; Decker, M. Probing the mid-gorge of cholinesterases with spacer-modified bivalent quinazolinimines leads to highly potent and selective butyrylcholinesterase inhibitors. Bioorg. Med. Chem. 2011, 19, 1222–1235. [Google Scholar] [CrossRef]
- Darras, F.H.; Wehle, S.; Huang, G.Z.; Sotriffer, C.A.; Decker, M. Amine substitution of quinazolinones leads to selective nanomolar AChE inhibitors with ‘inverted’ binding mode. Bioorg. Med. Chem. 2014, 22, 4867–4881. [Google Scholar] [CrossRef]
- Daoud, I.; Bouarab, S.; Ghalem, S. Docking, dynamic simulation and quantum mechanics studies of pyrazinamide derivatives as novel inhibitors of Acetylcholinesterase and Butyrylcholinesterase. Pharma Chem. 2015, 7, 307–321. [Google Scholar]
- Tanoli, N.U.; Tanoli, S.A.K.; Ferreira, A.G.; Mehmood, M.; Gul, S.; Monteiro, J.L.; Vieira, L.C.C.; Venancio, T.; Correa, A.G.; Ul-Haq, Z. Characterization of the interactions between coumarin-derivatives and acetylcholinesterase: Examination by NMR and docking simulations. J. Mol. Modeling 2018, 24, 11. [Google Scholar] [CrossRef]
- Saeed, A.; Zaib, S.; Ashraf, S.; Iftikhar, J.; Muddassar, M.; Zhang, K.Y.J.; Iqbal, J. Synthesis, cholinesterase inhibition and molecular modelling studies of coumarin linked thiourea derivatives. Bioorg. Chem. 2015, 63, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.L.; Fernandez-Nieto, F.; Brea, J.M.; Catto, M.; Paleo, M.R.; Porto, S.; Sardina, F.J.; Castro, M.; Pisani, L.; Carotti, A.; et al. 8-Aminomethyl-7-hydroxy-4-methylcoumarins as Multitarget Leads for Alzheimer’s Disease. ChemistrySelect 2016, 1, 2742–2749. [Google Scholar] [CrossRef]
- Abu-Aisheh, M.N.; Al-Aboudi, A.; Mustafa, M.S.; El-Abadelah, M.M.; Ali, S.Y.; Ul-Haq, Z.; Mubarak, M.S. Coumarin derivatives as acetyl- and butyrylcholinestrase inhibitors: An in vitro, molecular docking, and molecular dynamics simulations study. Heliyon 2019, 5, e01552. [Google Scholar] [CrossRef] [Green Version]
- Kwong, H.C.; Mah, S.H.; Chia, T.S.; Quah, C.K.; Lim, G.K.; Kumar, C.S.C. Cholinesterase Inhibitory Activities of Adamantyl-Based Derivatives and Their Molecular Docking Studies. Molecules 2017, 22, 1005. [Google Scholar] [CrossRef] [Green Version]
- Stoddard, S.V.; Hamann, M.T.; Wadkins, R.M. Insights and Ideas Garnered from Marine Metabolites for Development of Dual- Function Acetylcholinesterase and Amyloid-beta Aggregation Inhibitors. Mar. Drugs 2014, 12, 2114–2131. [Google Scholar] [CrossRef]
- Hassan, M.; Raza, H.; Abbasi, M.A.; Moustafa, A.A.; Seo, S.Y. The exploration of novel Alzheimer’s therapeutic agents from the pool of FDA approved medicines using drug repositioning, enzyme inhibition and kinetic mechanism approaches. Biomed. Pharmacother. 2019, 109, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Dalmizrak, O.; Terali, K.; Yetkin, O.; Ogus, I.H.; Ozer, N. Computational and experimental studies on the interaction between butyrylcholinesterase and fluoxetine: Implications in health and disease. Xenobiotica 2019, 49, 803–810. [Google Scholar] [CrossRef]
- Fang, J.; Pang, X.; Wu, P.; Yan, R.; Gao, L.; Li, C.; Lian, W.; Wang, Q.; Liu, A.L.; Du, G.H. Molecular Modeling on Berberine Derivatives toward BuChE: An Integrated Study with Quantitative Structure–Activity Relationships Models, Molecular Docking, and Molecular Dynamics Simulations. Chem. Biol. Drug Des. 2016, 87, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Ayupov, R.K.; Akberova, N.I. Molecular dynamics of the pyridoxine derivative in the acetylcholinesterase active cavity. Res. J. Pharm. Biol. Chem. Sci. 2015, 6, 1717–1722. [Google Scholar]
- Zhou, S.; Yuan, Y.X.; Zheng, F.; Zhan, C.G. Structure-based virtual screening leading to discovery of highly selective butyrylcholinesterase inhibitors with solanaceous alkaloid scaffolds. Chem. Biol. Interact. 2019, 308, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, Y.K.; Liu, Z.H.; Yang, L.; Tang, J.G.; Miao, M.M.; Gan, N.; Li, H. Differences between the binding modes of enantiomers S/R-nicotine to acetylcholinesterase. RSC Adv. 2019, 9, 1428–1440. [Google Scholar] [CrossRef] [Green Version]
- Terali, K. An evaluation of neonicotinoids’ potential to inhibit human cholinesterases: Protein-ligand docking and interaction profiling studies. J. Mol. Graph. Model. 2018, 84, 54–63. [Google Scholar] [CrossRef]
- Gao, D.; Zhan, C.G. Modeling evolution of hydrogen bonding and stabilization of transition states in the process of cocaine hydrolysis catalyzed by human butyrylcholinesterase. Proteins Struct. Funct. Bioinform. 2006, 62, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Hou, S.R.; Xue, L.; Zheng, F.; Zhan, C.G. Amino-acid mutations to extend the biological half-life of a therapeutically valuable mutant of human butyrylcholinesterase. Chem. Biol. Interact. 2014, 214, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Pang, Y.-P.; Lockridge, O.; Brimijoin, S. Re-engineering Butyrylcholinesterase as a Cocaine Hydrolase. Mol. Pharmacol. 2002, 62, 220. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Yang, W.; Ko, M.-C.; Liu, J.; Cho, H.; Gao, D.; Tong, M.; Tai, H.-H.; Woods, J.H.; Zhan, C.-G. Most Efficient Cocaine Hydrolase Designed by Virtual Screening of Transition States. J. Am. Chem. Soc. 2008, 130, 12148–12155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamza, A.; Cho, H.; Tai, H.-H.; Zhan, C.-G. Molecular Dynamics Simulation of Cocaine Binding with Human Butyrylcholinesterase and Its Mutants. J. Phys. Chem. B 2005, 109, 4776–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.; Ko, M.-C.; Tong, M.; Yang, W.; Hou, S.; Fang, L.; Liu, J.; Zheng, F.; Woods, J.H.; Tai, H.-H.; et al. Design, preparation, and characterization of high-activity mutants of human butyrylcholinesterase specific for detoxification of cocaine. Mol. Pharmacol. 2011, 79, 290–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Elena, A.; Nan, S.; James, D.P.; Jeffry, D.W.; Stephen, B. Gene Transfer of Cocaine Hydrolase Suppresses Cardiovascular Responses to Cocaine in Rats. Mol. Pharm. 2005, 67, 204–211. [Google Scholar]
- Chen, X.B.; Huang, X.Q.; Geng, L.Y.; Xue, L.; Hou, S.R.; Zheng, X.R.; Brimijoin, S.; Zheng, F.; Zhan, C.G. Kinetic characterization of a cocaine hydrolase engineered from mouse butyrylcholinesterase. Biochem. J. 2015, 466, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Moralev, S.N.; Tikhonov, D.B. Investigation of structure-activity relationships in organophosphates-cholinesterase interaction using docking analysis. Chem. Biol. Interact. 2010, 187, 153–156. [Google Scholar] [CrossRef]
- Veselinovic, J.B.; Nikolic, G.M.; Trutic, N.V.; Zivkovic, J.V.; Veselinovic, A.M. Monte Carlo QSAR models for predicting organophosphate inhibition of acetycholinesterase. Sar Qsar Environ. Res. 2015, 26, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, J.; Zheng, H.; Zhong, J.; Zhou, J. Simulated revelation of the adsorption behaviours of acetylcholinesterase on charged self-assembled monolayers. Nanoscale 2020, 12, 3701–3714. [Google Scholar] [CrossRef]
- Bondžić, A.M.; Lazarević-Pašti, T.D.; Leskovac, A.R.; Petrović, S.Ž.; Čolović, M.B.; Parac-Vogt, T.N.; Janjić, G.V. A new acetylcholinesterase allosteric site responsible for binding voluminous negatively charged molecules—The role in the mechanism of AChE inhibition. Eur. J. Pharm. Sci. 2020, 151. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Schwans, J.P.; Sorin, E.J.; Tran, T.; Gonzalez, J.; Arteaga, E.; McCoy, S.; Alvarado, W. Synthesis, biochemical evaluation, and molecular modeling studies of aryl and arylalkyl di-n-butyl phosphates, effective butyrylcholinesterase inhibitors. Bioorg. Med. Chem. 2017, 25, 3171–3181. [Google Scholar] [CrossRef]
- Sorin, E.J.; Alvarado, W.; Cao, S.; Radcliffe, A.; La, P.; An, Y. Ensemble molecular dynamics of a protein-ligand complex: Residual inhibitor entropy enhances drug potency in butyrylcholinesterase. Bioenergetics 2017, 6, 145. [Google Scholar] [CrossRef] [Green Version]
- Bremer, P.L.; De Boer, D.; Alvarado, W.; Martinez, X.; Sorin, E.J. Overcoming the Heuristic Nature of k -Means Clustering: Identification and Characterization of Binding Modes from Simulations of Molecular Recognition Complexes. J. Chem. Inf. Modeling 2020, 60, 3081–3092. [Google Scholar] [CrossRef] [PubMed]
- Carlacci, L.; Millard, C.B.; Olson, M.A. Conformational energy landscape of the acyl pocket loop in acetylcholinesterase: A Monte Carlo-generalized Born model study. Biophys. Chem. 2004, 111, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.; Javor, S.; Ryan, D.A.; Smith, E.M.; Wang, B.; Zhang, J.; Cashman, J.R. Novel Human Butyrylcholinesterase Variants: Toward Organophosphonate Detoxication. Biochemistry 2014, 53, 4476–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, P.; Lockridge, O. Butyrylcholinesterase for protection from organophosphorus poisons: Catalytic complexities and hysteretic behavior. Arch. Biochem. Biophys. 2010, 494, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, S.; Beck Jeremy, M.; Xia, S.; Zhang, J.; Hadad Christopher, M. Butyrylcholinesterase and G116H, G116S, G117H, G117N, E197Q and G117H/E197Q mutants: A molecular dynamics study. Chem. Biol. Interact. 2010, 187, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Nachon, F.; Carletti, E.; Wandhammer, M.; Nicolet, Y.; Schopfer, L.M.; Masson, P.; Lockridge, O. X-ray crystallographic snapshots of reaction intermediates in the G117H mutant of human butyrylcholinesterase, a nerve agent target engineered into a catalytic bioscavenger. Biochem. J. 2011, 434, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Amitay, M.; Shurki, A. The structure of G117H mutant of butyrylcholinesterase: Nerve agents scavenger. Proteins 2009, 77, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Liu, J.J.; Zhan, C.G. Why Does the G117H Mutation Considerably Improve the Activity of Human Butyrylcholinesterase against Sarin? Insights from Quantum Mechanical/Molecular Mechanical Free Energy Calculations. Biochemistry 2012, 51, 8980–8992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorecki, L.; Korabecny, J.; Musilek, K.; Malinak, D.; Nepovimova, E.; Dolezal, R.; Jun, D.; Soukup, O.; Kuca, K. SAR study to find optimal cholinesterase reactivator against organophosphorous nerve agents and pesticides. Arch. Toxicol. 2016, 90, 2831–2859. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, J.O.S.; Franca, T.C.C.; Kuca, K.; da Cunha, E.F.F.; Abagyan, R.; Mancini, D.T.; Ramalho, T.C. Molecular modeling and in vitro reactivation study between the oxime BI-6 and acetylcholinesterase inhibited by different nerve agents. J. Biomol. Struct. Dyn. 2015, 33, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Worek, F.; Aurbek, N.; Herkert, N.M.; John, H.; Eddleston, M.; Eyer, P.; Thiermann, H. Evaluation of medical countermeasures against organophosphorus compounds: The value of experimental data and computer simulations. Chem. Biol. Interact. 2010, 187, 259–264. [Google Scholar] [CrossRef]
- Allgardsson, A.; Berg, L.; Akfur, C.; Hörnberg, A.; Worek, F.; Linusson, A.; Ekström, F.J. Structure of a prereaction complex between the nerve agent sarin, its biological target acetylcholinesterase, and the antidote HI-6. Proc. Natl. Acad. Sci. USA 2016, 113, 5514–5519. [Google Scholar] [CrossRef] [Green Version]
- Veselinovic, A.M.; Veselinovic, J.B.; Toropov, A.A.; Toropov, A.P.; Nikolic, G.M. QSAR models for the reactivation of sarin inhibited acetylcholinesterase by quaternary pyridinium oximes based on Monte Carlo method. Curr. Comput. Aided Drug Des. 2014, 10, 266–273. [Google Scholar] [CrossRef]
- de Souza, F.R.; Garcia, D.R.; Cuya, T.; Pimentel, A.S.; Goncalves, A.D.; de Alencastro, R.B.; Franca, T.C.C. Molecular Modeling Study of Uncharged Oximes Compared to HI-6 and 2-PAM Inside Human AChE Sarin and VX Conjugates. ACS Omega 2020, 5, 4490–4500. [Google Scholar] [CrossRef]
- Musilek, K.; Roder, J.; Komloova, M.; Holas, O.; Hrabinova, M.; Pohanka, M.; Dohnal, V.; Opletalova, V.; Kuca, K.; Jung, Y.S. Preparation, in vitro screening and molecular modelling of symmetrical 4-tert-butylpyridinium cholinesterase inhibitors-Analogues of SAD-128. Bioorg. Med. Chem. Lett. 2011, 21, 150–154. [Google Scholar] [CrossRef]
- Bhattacharjee, A.K.; Kuca, K.; Musilek, K.; Gordon, R.K. In Silico Pharmacophore Model for Tabun-Inhibited Acetylcholinesterase Reactivators: A Study of Their Stereoelectronic Properties. Chem. Res. Toxicol. 2010, 23, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Chandar, N.B.; Lo, R.; Ganguly, B. Quantum chemical and steered molecular dynamics studies for one pot solution to reactivate aged acetylcholinesterase with alkylator oxime. Chem. Biol. Interact. 2014, 223, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Ganguly, B. Can hydroxylamine be a more potent nucleophile for the reactivation of tabun-inhibited AChE than prototype oxime drugs? An answer derived from quantum chemical and steered molecular dynamics studies. Mol. Biosyst. 2014, 10, 2368–2383. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.A.V.; Pereira, A.F.; LaPlante, S.R.; Kuca, K.; Ramalho, T.C.; Franca, T.C.C. Reactivation of VX-Inhibited Human Acetylcholinesterase by Deprotonated Pralidoxime. A Complementary Quantum Mechanical Study. Biomolecules 2020, 10, 192. [Google Scholar] [CrossRef] [Green Version]
- de Castro, A.A.; Polisel, D.A.; Pereira, B.T.L.; da Cunha, E.F.F.; Kuca, K.; Nepovimova, E.; Ramalho, T.C. Understanding the Interaction Modes and Reactivity of Trimedoxime toward MmAChE Inhibited by Nerve Agents: Theoretical and Experimental Aspects. Int. J. Mol. Sci. 2020, 21, 6510. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.-P.; Kollmeyer, T.M.; Hong, F.; Lee, J.-C.; Hammond, P.I.; Haugabouk, S.P.; Brimijoin, S. Rational Design of Alkylene-Linked Bis-Pyridiniumaldoximes as Improved Acetylcholinesterase Reactivators. Chem. Biol. 2003, 10, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Driant, T.; Nachon, F.; Ollivier, C.; Renard, P.Y.; Derat, E. On the Influence of the Protonation States of Active Site Residues on AChE Reactivation: A QM/MM Approach. Chembiochem 2017, 18, 666–675. [Google Scholar] [CrossRef]
- Malinak, D.; Dolezal, R.; Hepnarova, V.; Hozova, M.; Andrys, R.; Bzonek, P.; Racakova, V.; Korabecny, J.; Gorecki, L.; Mezeiova, E.; et al. Synthesis, in vitro screening and molecular docking of isoquinolinium-5-carbaldoximes as acetylcholinesterase and butyrylcholinesterase reactivators. J. Enzym. Inhib. Med. Chem. 2020, 35, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Vitorović-Todorović, M.D.; Worek, F.; Perdih, A.; Bauk, S.Đ.; Vujatović, T.B.; Cvijetić, I.N. The in vitro protective effects of the three novel nanomolar reversible inhibitors of human cholinesterases against irreversible inhibition by organophosphorous chemical warfare agents. Chem. Biol. Interact. 2019, 309, 108714. [Google Scholar] [CrossRef]
- Mesaric, T.; Baweja, L.; Drasler, B.; Drobne, D.; Makovec, D.; Dusak, P.; Dhawan, A.; Sepcic, K. Effects of surface curvature and surface characteristics of carbon-based nanomaterials on the adsorption and activity of acetylcholinesterase. Carbon 2013, 62, 222–232. [Google Scholar] [CrossRef]
- Vats, C.; Dhanjal, J.K.; Goyal, S.; Bharadvaja, N.; Grover, A. Computational design of novel flavonoid analogues as potential AChE inhibitors: Analysis using group-based QSAR, molecular docking and molecular dynamics simulations. Struct. Chem. 2015, 26, 467–476. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.H.; Lee, H.W.; Sun, Y.N.; Jang, W.H.; Yang, S.Y.; Jang, H.B.; Kim, Y.H. (-)-Epicatechin derivate from Orostachys japonicus as potential inhibitor of the human butyrylcholinesterase. Int. J. Biol. Macromol. 2016, 91, 1033–1039. [Google Scholar] [CrossRef]
- de Almeida, J.S.F.D.; Cavalcante, S.F.d.A.; Dolezal, R.; Kuca, K.; Musilek, K.; Jun, D.; Franca, T.C.C. Surface screening, molecular modeling and in vitro studies on the interactions of aflatoxin M1 and human enzymes acetyl- and butyrylcholinesterase. Chem. Biol. Interact. 2019, 308, 113–119. [Google Scholar] [CrossRef]
- Delogu, G.L.; Matos, M.J.; Fanti, M.; Era, B.; Medda, R.; Pieroni, E.; Fais, A.; Kumar, A.; Pintus, F. 2-Phenylbenzofuran derivatives as butyrylcholinesterase inhibitors: Synthesis, biological activity and molecular modeling. Bioorg. Med. Chem. Lett. 2016, 26, 2308–2313. [Google Scholar] [CrossRef] [PubMed]
- Estevez, J.; de Souza, F.R.; Romo, M.; Mangas, I.; Franca, T.C.C.; Vilanova, E. Interactions of human butyrylcholinesterase with phenylvalerate and acetylthiocholine as substrates and inhibitors: Kinetic and molecular modeling approaches. Arch. Toxicol. 2019, 93, 1281–1296. [Google Scholar] [CrossRef]
- Pourshojaei, Y.; Abiri, A.; Eskandari, K.; Haghighijoo, Z.; Edraki, N.; Asadipour, A. Phenoxyethyl Piperidine/Morpholine Derivatives as PAS and CAS Inhibitors of Cholinesterases: Insights for Future Drug Design. Sci. Rep. 2019, 9, 19855. [Google Scholar] [CrossRef]
- Hudcova, A.; Kroutil, A.; Kubinova, R.; Garro, A.D.; Gutierrez, L.J.; Enriz, D.; Oravec, M.; Csollei, J. Arylaminopropanone Derivatives as Potential Cholinesterase Inhibitors: Synthesis, Docking Study and Biological Evaluation. Molecules 2020, 25, 1751. [Google Scholar] [CrossRef]
- Gharaghani, S.; Khayamian, T.; Ebrahimi, M. Molecular dynamics simulation study and molecular docking descriptors in structure-based QSAR on acetylcholinesterase (AChE) inhibitors. Sar Qsar Environ. Res. 2013, 24, 773–794. [Google Scholar] [CrossRef]
- Khan, I.; Samad, A.; Khan, A.Z.; Habtemariam, S.; Badshah, A.; Abdullah, S.M.; Ullah, N.; Khan, A.; Zia-Ul-Haq, M. Molecular interactions of 4-acetoxy-plakinamine B with peripheral anionic and other catalytic subsites of the aromatic gorge of acetylcholinesterase: Computational and structural insights. Pharm. Biol. 2013, 51, 722–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, S.K.; Sinha, S.K.; Srivastava, P.; Tripathi, P.N.; Sharma, P.; Tripathi, M.K.; Tripathi, A.; Choubey, P.K.; Waiker, D.K.; Aggarwal, L.M.; et al. Design and development of novel p-aminobenzoic acid derivatives as potential cholinesterase inhibitors for the treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 82, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Coban, G.; Carlino, L.; Tarikogullari, A.H.; Parlar, S.; Sarikaya, G.; Alptuzun, V.; Alpan, A.S.; Gunes, H.S.; Erciyas, E. 1H-benzimidazole derivatives as butyrylcholinesterase inhibitors: Synthesis and molecular modeling studies. Med. Chem. Res. 2016, 25, 2005–2014. [Google Scholar] [CrossRef]
- Abuhamdah, S.; Habash, M.; Taha, M. Elaborate ligand-based modeling coupled with QSAR analysis and in silico screening reveal new potent acetylcholinesterase inhibitors. J. Comput. Aided Mol. Des. 2013, 27, 1075–1092. [Google Scholar] [CrossRef]
- Correa-Basurto, J.; Bello, M.; Rosales-Hernandez, M.C.; Hernandez-Rodriguez, M.; Nicolas-Vazquez, I.; Rojo-Dominguez, A.; Trujillo-Ferrara, J.G.; Miranda, R.; Flores-Sandoval, C.A. QSAR, docking, dynamic simulation and quantum mechanics studies to explore the recognition properties of cholinesterase binding sites. Chem. Biol. Interact. 2014, 209, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gwaram, N.S.; Ali, H.M.; Abdulla, M.A.; Buckle, M.J.C.; Sukumaran, S.D.; Chung, L.Y.; Othman, R.; Alhadi, A.A.; Yehye, W.A.; Hadi, A.H.A.; et al. Synthesis, Characterization, X-ray Crystallography, Acetyl Cholinesterase Inhibition and Antioxidant Activities of Some Novel Ketone Derivatives of Gallic Hydrazide-Derived Schiff Bases. Molecules 2012, 17, 2408–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, R.; Galeazzi, R.; Rosi, G.; Fiorini, R.; Pirisinu, I.; Ambrosini, A.; Zolese, G. Anandamide and its congeners inhibit human plasma butyrylcholinesterase. Possible new roles for these endocannabinoids? Biochimie 2011, 93, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Vitorovic-Todorovic, M.D.; Koukoulitsa, C.; Juranic, I.O.; Mandic, L.M.; Drakulic, B.J. Structural modifications of 4-aryl-4-oxo-2-aminylbutanamides and their acetyl- and butyrylcholinesterase inhibitory activity. Investigation of AChE-ligand interactions by docking calculations and molecular dynamics simulations. Eur. J. Med. Chem. 2014, 81, 158–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Gupta, D. Discovery of potential inhibitor against human acetylcholinesterase: A molecular docking and molecular dynamics investigation. Comput. Biol. Chem. 2017, 68, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, T.; Ghayeb, Y. Atomic insight into designed carbamate-based derivatives as acetylcholine esterase (AChE) inhibitors: A computational study by multiple molecular docking and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2018, 36, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Kurt, B.Z.; Gazioglu, I.; Dag, A.; Salmas, R.E.; Kayik, G.; Durdagi, S.; Sonmez, F. Synthesis, anticholinesterase activity and molecular modeling study of novel carbamate-substituted thymol/carvacrol derivatives. Bioorg. Med. Chem. 2017, 25, 1352–1363. [Google Scholar] [CrossRef]
- Domínguez, J.L.; Fernández-Nieto, F.; Castro, M.; Catto, M.; Paleo, M.R.; Porto, S.; Sardina, F.J.; Brea, J.M.; Carotti, A.; Villaverde, M.C.; et al. Computer-Aided Structure-Based Design of Multitarget Leads for Alzheimer’s Disease. J. Chem. Inf. Modeling 2015, 55, 135–148. [Google Scholar] [CrossRef]
- Dileep, K.V.; Remya, C.; Tintu, I.; Sadasivan, C. Inhibition, ADME and structure based modification of IAA and IBA against acetylcholinesterase: An attempt towards new drug development for Alzheimer’s disease. Front. Life Sci. 2013, 7, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Bingul, M.; Ercan, S.; Boga, M. The design of novel 4,6-dimethoxyindole based hydrazide- hydrazones: Molecular modeling, synthesis and anticholinesterase activity. J. Mol. Struct. 2020, 1213, 128202. [Google Scholar] [CrossRef]
- Abdul Manap, A.S.; Tan, A.C.W.; Weng, H.L.; Chia, A.Y.Y.; Vijayabalan, S.; Arya, A.; Wong, E.H.; Rizwan, F.; Bindal, U.; Koshy, S.; et al. Synergistic Effects of Curcumin and Piperine as Potent Acetylcholine and Amyloidogenic Inhibitors With Significant Neuroprotective Activity in SH-SY5Y Cells via Computational Molecular Modeling and in vitro Assay. Front. Aging Neurosci. 2019, 11, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meena, P.; Nemaysh, V.; Khatri, M.; Manral, A.; Luthra, P.M.; Tiwari, M. Synthesis, biological evaluation and molecular docking study of novel piperidine and piperazine derivatives as multi-targeted agents to treat Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 1135–1148. [Google Scholar] [CrossRef]
- Tripathi, P.N.; Srivastava, P.; Sharma, P.; Tripathi, M.K.; Seth, A.; Tripathi, A.; Rai, S.N.; Singh, S.P.; Shrivastava, S.K. Biphenyl-3-oxo-1,2,4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory. Bioorg. Chem. 2019, 85, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Gurung, A.B.; Aguan, K.; Mitra, S.; Bhattacharjee, A. Identification of molecular descriptors for design of novel Isoalloxazine derivatives as potential Acetylcholinesterase inhibitors against Alzheimer’s disease. J. Biomol. Struct. Dyn. 2017, 35, 1729–1742. [Google Scholar] [CrossRef]
- Sinha, S.K.; Shrivastava, S.K. Synthesis, evaluation and molecular dynamics study of some new 4-aminopyridine semicarbazones as an antiamnesic and cognition enhancing agents. Bioorg. Med. Chem. 2013, 21, 5451–5460. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.V.; Patel, N.R.; Kanhed, A.M.; Teli, D.M.; Patel, K.B.; Joshi, P.D.; Patel, S.P.; Gandhi, P.M.; Chaudhary, B.N.; Prajapati, N.K.; et al. Novel carbazole-stilbene hybrids as multifunctional anti-Alzheimer agents. Bioorg. Chem. 2020, 101. [Google Scholar] [CrossRef]
- Tallini, L.R.; Bastida, J.; Cortes, N.; Osorio, E.H.; Theoduloz, C.; Schmeda-Hirschmann, G. Cholinesterase Inhibition Activity, Alkaloid Profiling and Molecular Docking of Chilean Rhodophiala (Amaryllidaceae). Molecules 2018, 23, 1532. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, Y.A.; Gutierrez, M.; Ramirez, D.; Alzate-Morales, J.; Bernal, C.C.; Guiza, F.M.; Bohorquez, A.R.R. Novel N-allyl/propargyl tetrahydroquinolines: Synthesis via Three-component Cationic Imino Diels-Alder Reaction, Binding Prediction, and Evaluation as Cholinesterase Inhibitors. Chem. Biol. Drug Des. 2016, 88, 498–510. [Google Scholar] [CrossRef]
- Rodriguez Nunez, Y.A.; Gutierrez, M.; Alzate-Morales, J.; Adasme-Carreno, F.; Guiza, F.M.; Bernal, C.C.; Bohorquez, A.R.R. Tetrahydroquinoline-Isoxazole/Isoxazoline Hybrid Compounds as Potential Cholinesterases Inhibitors: Synthesis, Enzyme Inhibition Assays, and Molecular Modeling Studies. Int. J. Mol. Sci. 2020, 21, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darras, F.H.; Pockes, S.; Huang, G.; Wehle, S.; Strasser, A.; Wittmann, H.-J.; Nimczick, M.; Sotriffer, C.A.; Decker, M. Synthesis, Biological Evaluation, and Computational Studies of Tri- and Tetracyclic Nitrogen-Bridgehead Compounds as Potent Dual-Acting AChE Inhibitors and hH3 Receptor Antagonists. ACS Chem. Neurosci. 2014, 5, 225–242. [Google Scholar] [CrossRef] [Green Version]
- Samadi, A.; de los Ríos, C.; Bolea, I.; Chioua, M.; Iriepa, I.; Moraleda, I.; Bartolini, M.; Andrisano, V.; Gálvez, E.; Valderas, C.; et al. Multipotent MAO and cholinesterase inhibitors for the treatment of Alzheimer’s disease: Synthesis, pharmacological analysis and molecular modeling of heterocyclic substituted alkyl and cycloalkyl propargyl amine. Eur. J. Med. Chem. 2012, 52, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Dolles, D.; Hoffmann, M.; Gunesch, S.; Marinelli, O.; Moeller, J.; Santoni, G.; Chatonnet, A.; Lohse, M.J.; Wittmann, H.-J.; Strasser, A.; et al. Structure-Activity Relationships and Computational Investigations into the Development of Potent and Balanced Dual-Acting Butyrylcholinesterase Inhibitors and Human Cannabinoid Receptor 2 Ligands with Pro-Cognitive in Vivo Profiles. J. Med. Chem. 2018, 61, 1646–1663. [Google Scholar] [CrossRef]
- Hassan, M.; Abbasi, M.A.; Rehman, A.U.; Siddiqui, S.Z.; Hussain, G.; Shah, S.A.A.; Shahid, M.; Seo, S.-Y. Exploration of synthetic multifunctional amides as new therapeutic agents for Alzheimer’s disease through enzyme inhibition, chemoinformatic properties, molecular docking and dynamic simulation insights. J. Theor. Biol. 2018, 458, 169–183. [Google Scholar] [CrossRef]
- Hassan, M.; Abbasi, M.A.; Rehman, A.U.; Siddiqui, S.Z.; Shahzadi, S.; Raza, H.; Hussain, G.; Ali Shah, S.A.; Ashraf, M.; Shahid, M.; et al. Designing of promising medicinal scaffolds for Alzheimer’s disease through enzyme inhibition, lead optimization, molecular docking and dynamic simulation approaches. Bioorg. Chem. 2019, 91, 103138. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Du, C.; Li, Q.; Chen, T.; Lu, X.; Li, Q.; Feng, F.; Chen, Y.; Liu, W.; Sun, H. Discovery, molecular dynamic simulation and biological evaluation of structurally diverse cholinesterase inhibitors with new scaffold through shape-based pharmacophore virtual screening. Bioorg. Chem. 2019, 92, 103294. [Google Scholar] [CrossRef]
- Asadi, M.; Ebrahimi, M.; Mohammadi-Khanaposhtani, M.; Azizian, H.; Sepehri, S.; Nadri, H.; Biglar, M.; Amanlou, M.; Larijani, B.; Mirzazadeh, R.; et al. Design, synthesis, molecular docking, and cholinesterase inhibitory potential of phthalimide-dithiocarbamate hybrids as new agents for treatment of Alzheimer’s disease. Chem. Biodivers. 2019, 16, e1900370. [Google Scholar] [CrossRef]
- Liu, S.J.; Shang, R.F.; Shi, L.X.; Zhou, R.; He, J.Y.; Wan, D.C.C. Design, Synthesis, and Evaluation of 7H-thiazolo-3,2-b-1,2,4-triazin-7-one Derivatives as Dual Binding Site Acetylcholinesterase Inhibitors. Chem. Biol. Drug Des. 2014, 84, 169–174. [Google Scholar] [CrossRef]
- Islam, M.M.; Rohman, M.A.; Gurung, A.B.; Bhattacharjee, A.; Aguan, K.; Mitra, S. Correlation of cholinergic drug induced quenching of acetylcholinesterase bound thioflavin-T fluorescence with their inhibition activity. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 189, 250–257. [Google Scholar] [CrossRef]
- Tai, K.; Shen, T.; Henchman, R.H.; Bourne, Y.; Marchot, P.; McCammon, J.A. Mechanism of Acetylcholinesterase Inhibition by Fasciculin: A 5-ns Molecular Dynamics Simulation. J. Am. Chem. Soc. 2002, 124, 6153–6161. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.H.; Kleywegt, G.J.; Ravelli, R.B.; Silman, I.; Sussman, J.L. Crystal structure of an acetylcholinesterase-fasciculin complex: Interaction of a three-fingered toxin from snake venom with its target. Structure 1995, 3, 1355–1366. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, Y.; Lazar, P.; Son, M.; Baek, A.; Thangapandian, S.; Jeong, N.Y.; Yoo, Y.H.; Lee, K.W. Binding conformation prediction between human acetylcholinesterase and cytochrome c using molecular modeling methods. J. Mol. Graph. Model. 2011, 29, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Sohail, I.; Rashid, S. Molecular Dynamics and Regulation of Butyrylcholinesterase Cholinergic Activity by RNA Binding Proteins. CNS Neurol. Disord. Drug Targets 2014, 13, 1366–1377. [Google Scholar] [CrossRef]
- Sohail, S. In Silico Study of miR-132 with mRNA of Acetylcholinesterase to Investigate the Binding Affinity for Interaction. J. Mol. Imaging Dyn. 2018, 8. [Google Scholar] [CrossRef]
- Kumar, B.; Dwivedi, A.R.; Sarkar, B.; Gupta, S.K.; Krishnamurthy, S.; Mantha, A.K.; Parkash, J.; Kumar, V. 4,6-Diphenylpyrimidine Derivatives as Dual Inhibitors of Monoamine Oxidase and Acetylcholinesterase for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Kumar, V.; Prashar, V.; Saini, S.; Dwivedi, A.R.; Bajaj, B.; Mehta, D.; Parkash, J.; Kumar, V. Dipropargyl substituted diphenylpyrimidines as dual inhibitors of monoamine oxidase and acetylcholinesterase. Eur. J. Med. Chem. 2019, 177, 221–234. [Google Scholar] [CrossRef]
- Cavdar, H.; Senturk, M.; Guney, M.; Durdagi, S.; Kayik, G.; Supuran, C.T.; Ekinci, D. Inhibition of acetylcholinesterase and butyrylcholinesterase with uracil derivatives: Kinetic and computational studies. J. Enzym. Inhib. Med. Chem. 2019, 34, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zueva, I.; Dias, J.; Lushchekina, S.; Semenov, V.; Mukhamedyarov, M.; Pashirova, T.; Babaev, V.; Nachon, F.; Petrova, N.; Nurullin, L.; et al. New evidence for dual binding site inhibitors of acetylcholinesterase as improved drugs for treatment of Alzheimer’s disease. Neuropharmacology 2019, 155, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Yigit, M.; Yigit, B.; Taslimi, P.; Ozdemir, I.; Karaman, M.; Gulcin, I. Novel amine-functionalized benzimidazolium salts: Synthesis, characterization, bioactivity, and molecular docking studies. J. Mol. Struct. 2020, 1207, 127802. [Google Scholar] [CrossRef]
- Bocca, C.C.; Rittner, R.; Hoehr, N.F.; Pinheiro, G.M.S.; Abiko, L.A.; Basso, E.A. Molecular modeling and biological evaluation of 2-N,N-dimethylaminecyclohexyl 1-N′N′-dimethylcarbamate isomers and their methylsulfate salts as cholinesterases inhibitors. J. Mol. Struct. 2010, 983, 194–199. [Google Scholar] [CrossRef]
- Kumar, A.; Tiwari, A.; Sharma, A. Changing Paradigm from one Target one Ligand Towards Multi-target Directed Ligand Design for Key Drug Targets of Alzheimer Disease: An Important Role of In Silico Methods in Multi-target Directed Ligands Design. Curr. Neuropharmacol. 2018, 16, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.H.; Wu, J.W.; Liu, H.L.; Zhao, J.H.; Liu, K.T.; Chuang, C.K.; Lin, H.Y.; Tsai, W.B.; Ho, Y. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Yang, H.; Li, Q.; Chen, Y.; Li, Q.; Zhou, Y.; Feng, F.; Liu, W.; Guo, Q.; Sun, H. Expansion of the scaffold diversity for the development of highly selective butyrylcholinesterase (BChE) inhibitors: Discovery of new hits through the pharmacophore model generation, virtual screening and molecular dynamics simulation. Bioorg. Chem. 2019, 85, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, H.; Yang, H.; Tan, R.; Bian, Y.; Fu, T.; Li, W.; Wu, L.; Pei, Y.; Sun, H. Discovery of new acetylcholinesterase and butyrylcholinesterase inhibitors through structure-based virtual screening. RSC Adv. 2017, 7, 3429–3438. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, R.; Rodriguez, C.A.; Zuluaga, A.F. Prediction of Ligands Binding Acetylcholinesterase with Potential Antidotal Activity: A Virtual Screening Approach. Mol. Inform. 2019, 38, 1800126. [Google Scholar] [CrossRef]
- Brus, B.; Kosak, U.; Turk, S.; Pislar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J.-P.; Gobec, S. Discovery, Biological Evaluation, and Crystal Structure of a Novel Nanomolar Selective Butyrylcholinesterase Inhibitor. J. Med. Chem. 2014, 57, 8167–8179. [Google Scholar] [CrossRef]
- Kosak, U.; Brus, B.; Knez, D.; Zakelj, S.; Trontelj, J.; Pislar, A.; Sink, R.; Jukič, M.; Zivin, M.; Podkowa, A.; et al. The Magic of Crystal Structure-Based Inhibitor Optimization: Development of a Butyrylcholinesterase Inhibitor with Picomolar Affinity and in Vivo Activity. J. Med. Chem 2018, 61, 119–139. [Google Scholar] [CrossRef]
- Fang, J.; Yang, R.; Gao, L.; Zhou, D.; Yang, S.; Liu, A.-l.; Du, G.-h. Predictions of BuChE Inhibitors Using Support Vector Machine and Naive Bayesian Classification Techniques in Drug Discovery. J. Chem. Inf. Modeling 2013, 53, 3009–3020. [Google Scholar] [CrossRef]
- Sakkiah, S.; Lee, K.W. Pharmacophore-based virtual screening and density functional theory approach to identifying novel butyrylcholinesterase inhibitors. Acta Pharmacol. Sin. 2012, 33, 964–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.Q.; Mu, Y.S.; Wu, F.C. An enantiomer-based virtual screening approach: Discovery of chiral organophosphates as acetyl cholinesterase inhibitors. Ecotoxicol. Environ. Saf. 2017, 138, 215–222. [Google Scholar] [CrossRef]
- Toropova, M.A.; Raska, I.; Raskova, M.; Toropov, A.A. The Utilization of the Monte Carlo Technique for Rational Drug Discovery. Comb. Chem. High Throughput Screen. 2016, 19, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Thai, N.Q.; Nguyen, H.L.; Linh, H.Q.; Li, M.S. Protocol for fast screening of multi-target drug candidates: Application to Alzheimer’s disease. J. Mol. Graph. Model. 2017, 77, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; Mazzolari, A.; Testa, B.; Pedretti, A. Binding Space Concept: A New Approach To Enhance the Reliability of Docking Scores and Its Application to Predicting Butyrylcholinesterase Hydrolytic Activity. J. Chem. Inf. Modeling 2017, 57, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Green, L.G.; Radic, Z.; Taylor, P.; Sharpless, K.B.; Olson, A.J.; Grynszpan, F. Automated Docking with Protein Flexibility in the Design of Femtomolar “Click Chemistry” Inhibitors of Acetylcholinesterase. J. Chem. Inf. Modeling 2013, 53, 898–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jiang, H.; Li, H. SHAFTS: A Hybrid Approach for 3D Molecular Similarity Calculation. 1. Method and Assessment of Virtual Screening. J. Chem. Inf. Modeling 2011, 51, 2372–2385. [Google Scholar] [CrossRef] [PubMed]
- Lešnik, S.; Štular, T.; Brus, B.; Knez, D.; Gobec, S.; Janežič, D.; Konc, J. LiSiCA: A Software for Ligand-Based Virtual Screening and Its Application for the Discovery of Butyrylcholinesterase Inhibitors. J. Chem. Inf. Modeling 2015, 55, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.Y.; Itai, A. Efficient method for high-throughput virtual screening based on flexible docking: Discovery of novel acetylcholinesterase inhibitors. J. Med. Chem. 2004, 47, 4818–4828. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Boer, D.; Nguyen, N.; Mao, J.; Moore, J.; Sorin, E.J. A Comprehensive Review of Cholinesterase Modeling and Simulation. Biomolecules 2021, 11, 580. https://doi.org/10.3390/biom11040580
De Boer D, Nguyen N, Mao J, Moore J, Sorin EJ. A Comprehensive Review of Cholinesterase Modeling and Simulation. Biomolecules. 2021; 11(4):580. https://doi.org/10.3390/biom11040580
Chicago/Turabian StyleDe Boer, Danna, Nguyet Nguyen, Jia Mao, Jessica Moore, and Eric J. Sorin. 2021. "A Comprehensive Review of Cholinesterase Modeling and Simulation" Biomolecules 11, no. 4: 580. https://doi.org/10.3390/biom11040580
APA StyleDe Boer, D., Nguyen, N., Mao, J., Moore, J., & Sorin, E. J. (2021). A Comprehensive Review of Cholinesterase Modeling and Simulation. Biomolecules, 11(4), 580. https://doi.org/10.3390/biom11040580