
EPHA3 Contributes to Epigenetic Suppression of PTEN in Radioresistant Head and Neck Cancer

Abstract
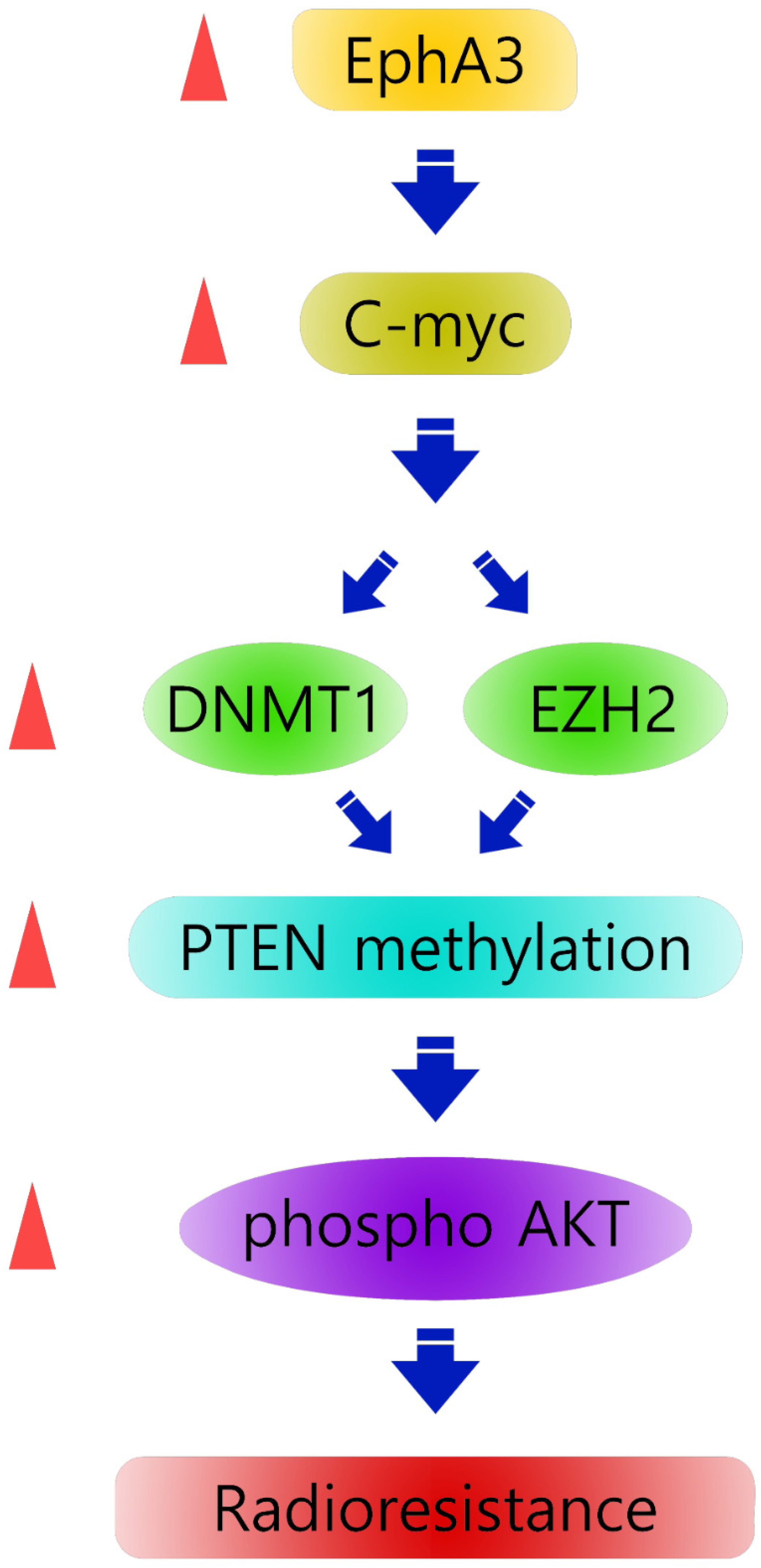
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Establishment of Radioresistant Head and Neck Cancer Cells and Reagents
2.2. Transfection of EPHA3 CRISPR/dCas9 Activation Plasmids into Cultured HN3-AMC Cells
2.3. RNA Interference
2.4. RNA Extraction and Quantitative Real-Time PCR Assay
2.5. Western Blotting Analysis
2.6. Bisulfite Modification and Methylation Specific PCR (MSP) Assay
- Unmethylated reaction, sense primer, 5′-GTGTTGGTGGAGGTAGTTGTTT-3′
- Antisense primer, 5′-ACCACTTAACTCTAAACCACAACCA-3′
- Methylated reaction, sense primer, 5′-TTCGTTCGTCGTCGTCGTATTT-3′
- Antisense primer, 5′-GCCGCTTAACTCTAAACCGCAACCG-3′.
2.7. Immunoprecipitation (IP)
2.8. Chromatin Immunoprecipitation (ChIP) Assays
2.9. Immunohistochemical Analysis
3. Results
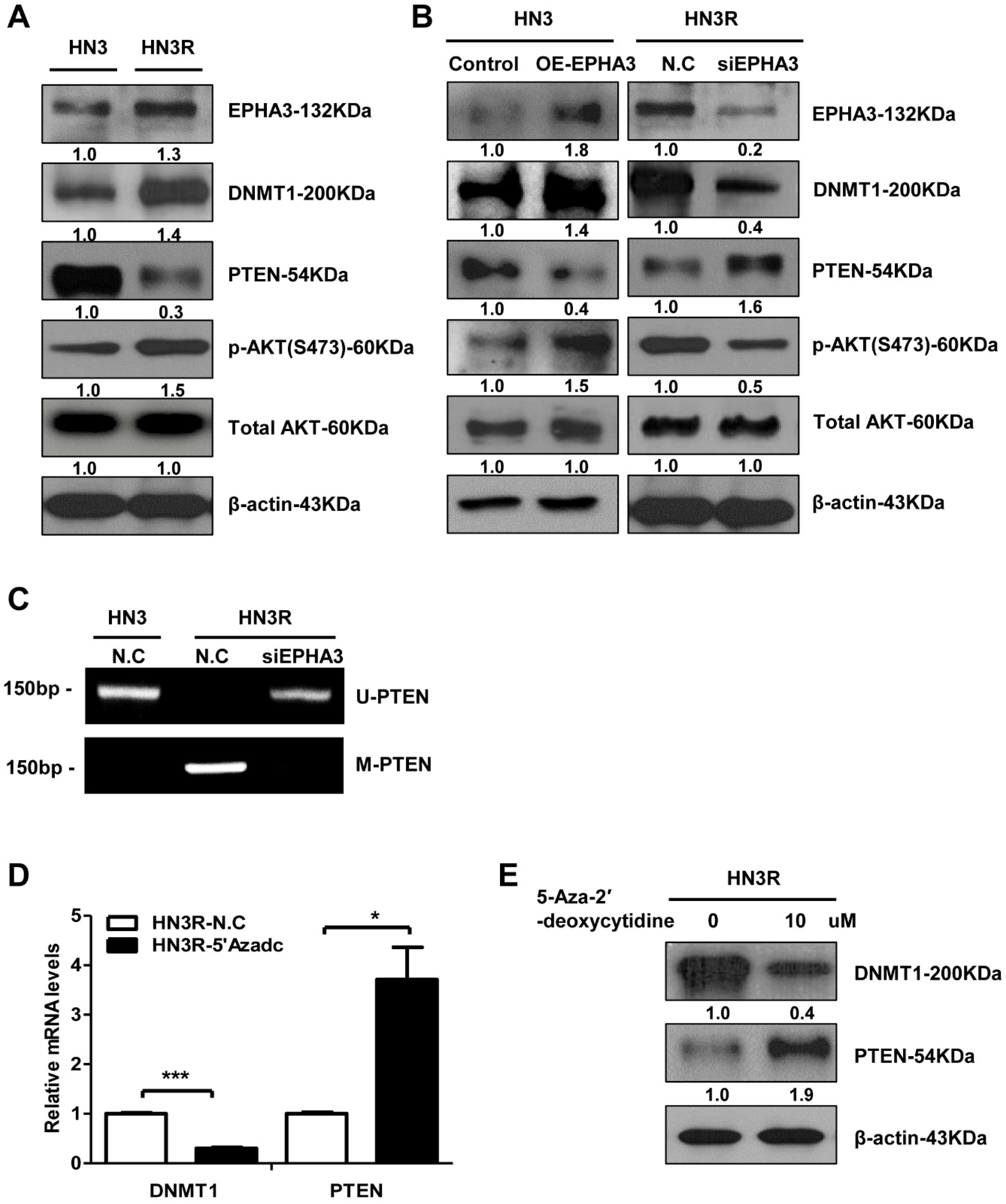
3.1. EPHA3 Maintains Sustained PTEN Suppression and Akt Activation through DNMT1-Mediated DNA Methylation in Radioresistant Head and Neck Cancer Cells
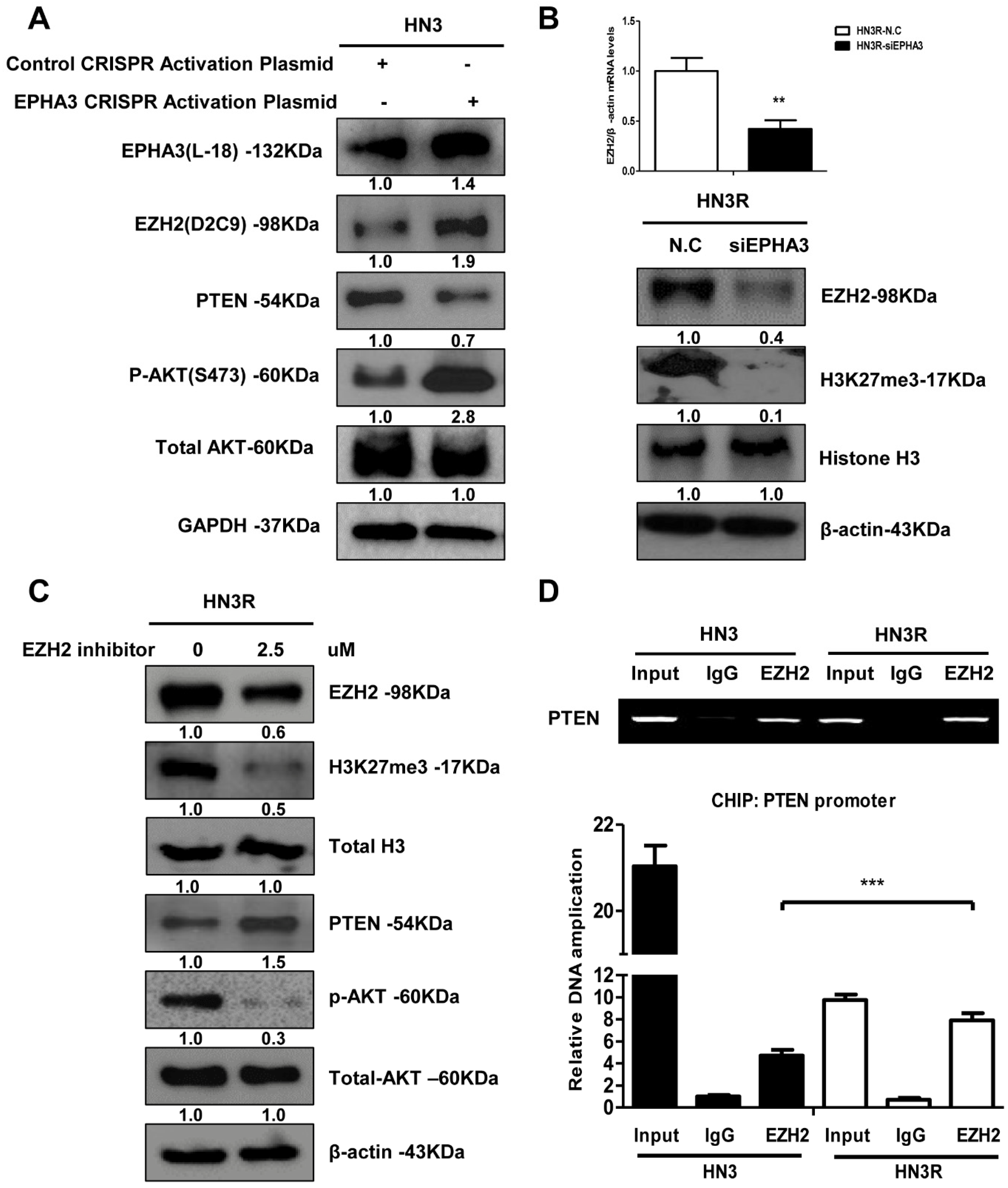
3.2. EPHA3 Maintains PTEN Suppression and Akt Activation through EZH2-Mediated Histone Methylation in Radioresistant Head and Neck Cancer Cells
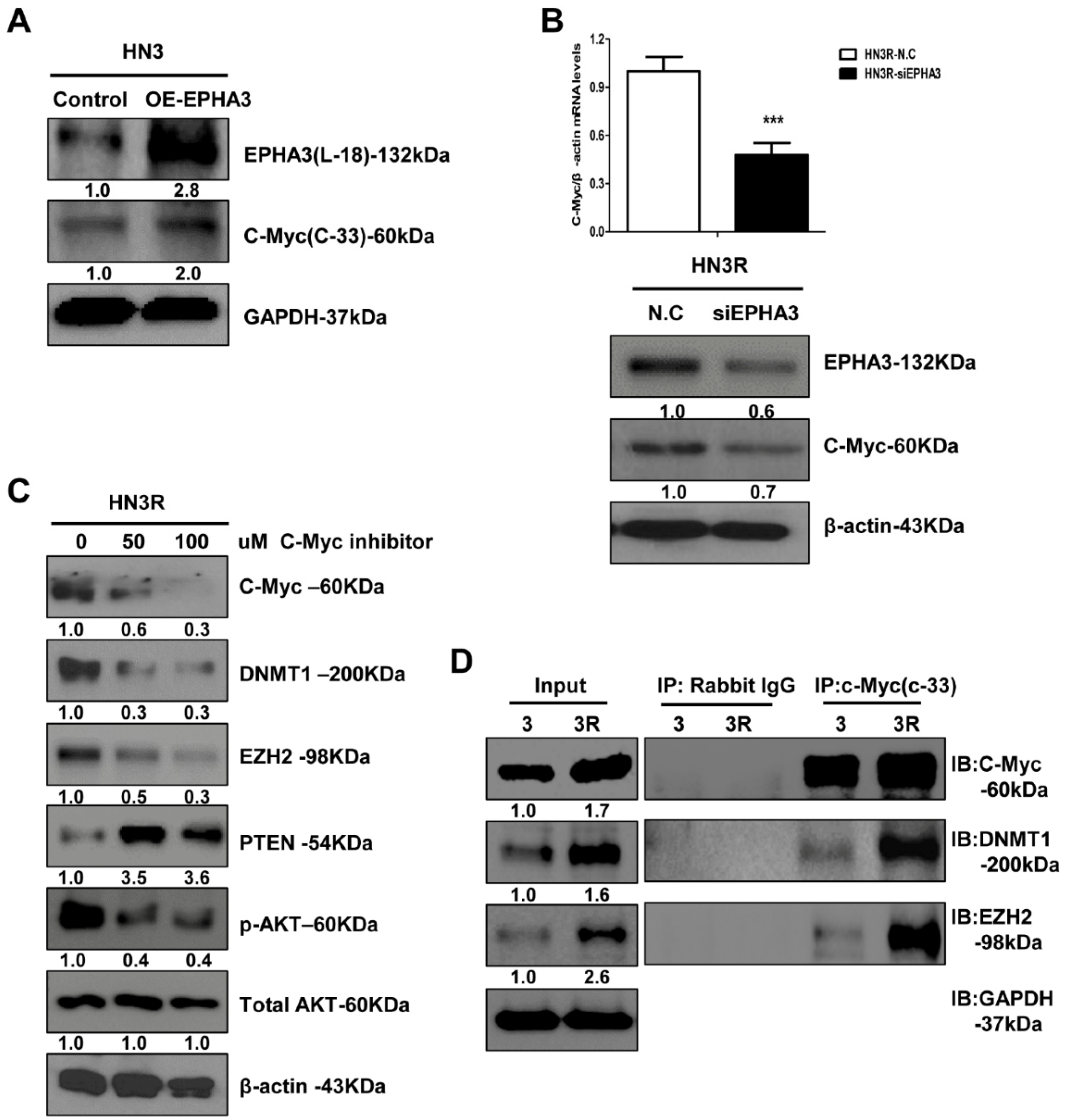
3.3. C-Myc Could Mediate the Functional Link between EPHA3 and DNMT1/EZH2 That Regulates PTEN-Based Radiation Resistance Epigenetically
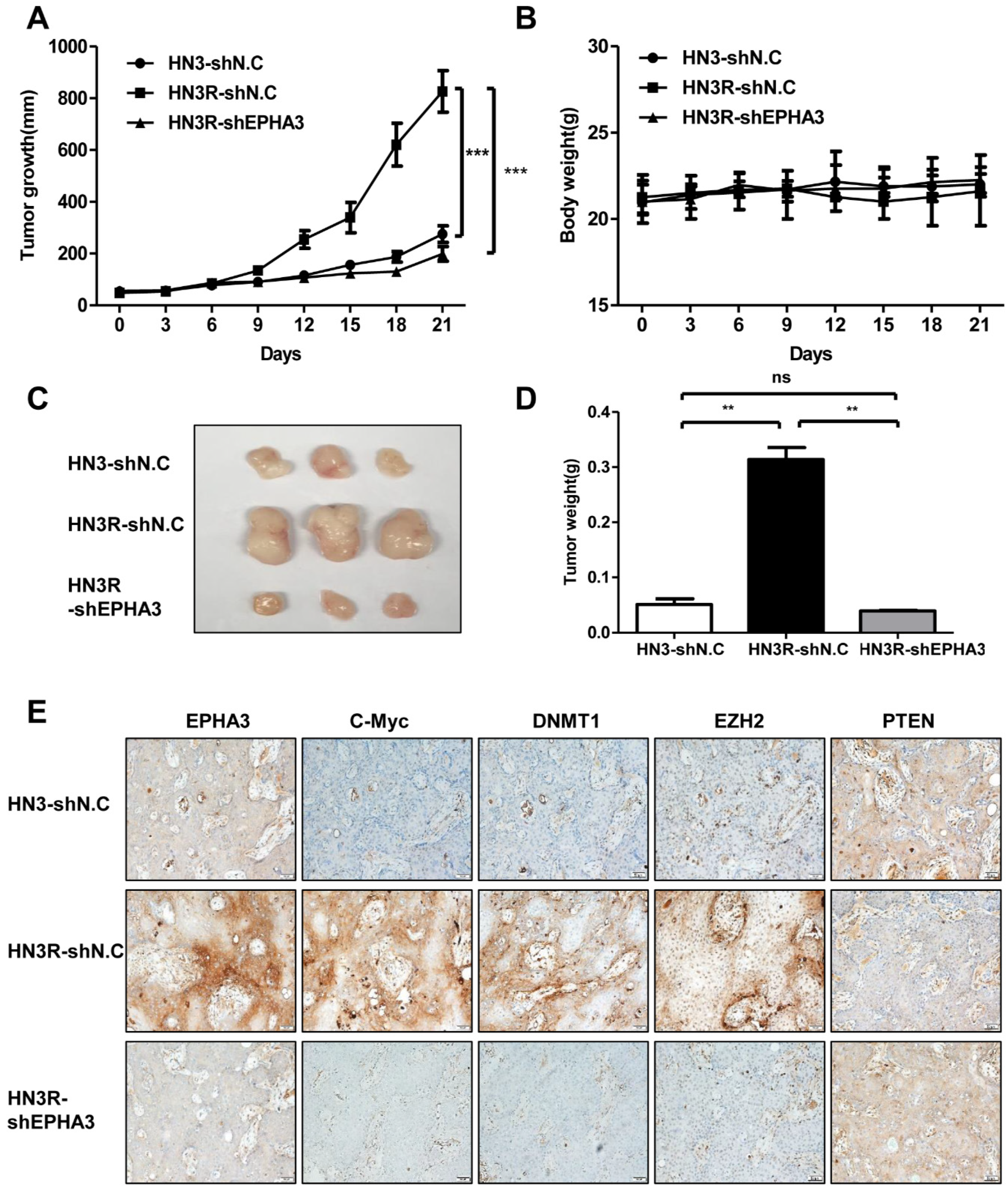
3.4. EPHA3 Inhibition Suppresses Tumor Growth through c-Myc, DNMT1, EZH2, and PTEN in an AMC HN3R Xenografts Model
3.5. EphA3 and c-Myc Is Overexpressed and PTEN Expression Is Decreased in Recurrent Laryngeal Cancer Specimens
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, K.K.; Toker, A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Rodemann, H.P. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin. Cancer Biol. 2015, 35, 180–190. [Google Scholar] [CrossRef]
- Luongo, F.; Colonna, F.; Calapa, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers 2019, 11, 1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, A.; Pandolfi, P.P. The PTEN(-)PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Leslie, N.R.; Downes, C.P. PTEN function: How normal cells control it and tumour cells lose it. Biochem. J. 2004, 382, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Ali-Hemmati, A.; Rahmati, M.; Charoudeh, H.N. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Lo, K.W.; Yim, S.F.; Chan, L.K.; Heung, M.S.; Chan, C.S.; Cheung, A.Y.; Chung, T.K.; Wong, Y.F. Epigenetic and genetic alternation of PTEN in cervical neoplasm. Gynecol. Oncol. 2004, 93, 621–627. [Google Scholar] [CrossRef]
- Garcia, J.M.; Silva, J.; Pena, C.; Garcia, V.; Rodriguez, R.; Cruz, M.A.; Cantos, B.; Provencio, M.; Espana, P.; Bonilla, F. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer 2004, 41, 117–124. [Google Scholar] [CrossRef]
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004, 64, 3014–3021. [Google Scholar] [CrossRef] [Green Version]
- Phuong, N.T.; Kim, S.K.; Lim, S.C.; Kim, H.S.; Kim, T.H.; Lee, K.Y.; Ahn, S.G.; Yoon, J.H.; Kang, K.W. Role of PTEN promoter methylation in tamoxifen-resistant breast cancer cells. Breast Cancer Res. Treat. 2011, 130, 73–83. [Google Scholar] [CrossRef]
- Maeda, M.; Murakami, Y.; Watari, K.; Kuwano, M.; Izumi, H.; Ono, M. CpG hypermethylation contributes to decreased expression of PTEN during acquired resistance to gefitinib in human lung cancer cell lines. Lung Cancer 2015, 87, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Ling, Y.; Zhu, M.; Zhou, L.; Wan, M.; Bao, Y.; Liu, Y. Promoter region methylation and loss of protein expression of PTEN and significance in cervical cancer. Biomed. Rep. 2014, 2, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Lee, W.H.; Kim, S.W.; Je, H.U.; Lee, J.C.; Chang, H.W.; Kim, Y.M.; Kim, K.; Kim, S.Y.; Han, M.W. EphA3 maintains radioresistance in head and neck cancers through epithelial mesenchymal transition. Cell. Signal. 2018, 47, 122–130. [Google Scholar] [CrossRef] [PubMed]
- London, M.; Gallo, E. Critical role of EphA3 in cancer and current state of EphA3 drug therapeutics. Mol. Biol. Rep. 2020, 47, 5523–5533. [Google Scholar] [CrossRef]
- Song, Z.; Gao, S.; Liu, Y.M.; Wang, Y.; Sun, Z.X.; Bao, D.; Liu, C. EphA3 promotes the proliferation of NPC cells through negatively regulating the ability of FOG2. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6735–6743. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.-Q.; Wu, X.-S.; Wei, B.; Chen, L. Aberrant expression of EphA3 in gastric carcinoma: Correlation with tumor angiogenesis and survival. J. Gastroenterol. 2012, 47, 785–794. [Google Scholar] [CrossRef]
- Duan, X.; Xu, X.; Yin, B.; Hong, B.; Liu, W.; Liu, Q.; Tao, Z. The prognosis value of EphA3 and the androgen receptor in prostate cancer treated with radical prostatectomy. J. Clin. Lab. Anal. 2019, 33, e22871. [Google Scholar] [CrossRef] [PubMed]
- Lahtela, J.; Pradhan, B.; Närhi, K.; Hemmes, A.; Särkioja, M.; Kovanen, P.E.; Brown, A.; Verschuren, E.W. The putative tumor suppressor gene EphA3 fails to demonstrate a crucial role in murine lung tumorigenesis or morphogenesis. Dis. Models Mech. 2015, 8, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Andretta, E.; Cartón-García, F.; Martínez-Barriocanal, Á.; de Marcondes, P.G.; Jimenez-Flores, L.M.; Macaya, I.; Bazzocco, S.; Bilic, J.; Rodrigues, P.; Nieto, R.; et al. Investigation of the role of tyrosine kinase receptor EPHA3 in colorectal cancer. Sci. Rep. 2017, 7, 41576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Liu, L.; Mims, J.; Punska, E.C.; Williams, K.E.; Zhao, W.; Arcaro, K.F.; Tsang, A.W.; Zhou, X.; Furdui, C.M. Analysis of DNA methylation and gene expression in radiation-resistant head and neck tumors. Epigenetics 2015, 10, 545–561. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Nie, Q.; Lan, J.; Ge, J.; Qiu, Y.; Mao, Q. C-Myc negatively controls the tumor suppressor PTEN by upregulating miR-26a in glioblastoma multiforme cells. Biochem. Biophys. Res. Commun. 2013, 441, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Deplus, R.; Didelot, C.; Loriot, A.; Viré, E.; De Smet, C.; Gutierrez, A.; Danovi, D.; Bernard, D.; Boon, T.; et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005, 24, 336–346. [Google Scholar] [CrossRef]
- Fu, X.; Wu, X.; Djekidel, M.N.; Zhang, Y. Myc and Dnmt1 impede the pluripotent to totipotent state transition in embryonic stem cells. Nat. Cell Biol. 2019, 21, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Kalkat, M.; Resetca, D.; Lourenco, C.; Chan, P.K.; Wei, Y.; Shiah, Y.J.; Vitkin, N.; Tong, Y.; Sunnerhagen, M.; Done, S.J.; et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72, 836–848.e837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskus, W.; Buckley, K.; Rao, R.; Mandawat, A.; Yang, Y.; Joshi, R.; Wang, Y.; Balusu, R.; Chen, J.; Koul, S.; et al. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer Biol. Ther. 2009, 8, 939–950. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; He, Q.; Wen, X.; Hong, X.; Yang, X.; Tang, X.; Zhang, P.; Lei, Y.; Sun, Y.; Zhang, J.; et al. EZH2-DNMT1-mediated epigenetic silencing of miR-142-3p promotes metastasis through targeting ZEB2 in nasopharyngeal carcinoma. Cell Death Differ. 2019, 26, 1089–1106. [Google Scholar] [CrossRef]
- Ning, X.; Shi, Z.; Liu, X.; Zhang, A.; Han, L.; Jiang, K.; Kang, C.; Zhang, Q. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett. 2015, 359, 198–205. [Google Scholar] [CrossRef]
- Lee, J.C.; Lee, W.H.; Min, Y.J.; Cha, H.J.; Han, M.W.; Chang, H.W.; Kim, S.A.; Choi, S.H.; Kim, S.W.; Kim, S.Y. Development of TRAIL resistance by radiation-induced hypermethylation of DR4 CpG island in recurrent laryngeal squamous cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 1203–1211. [Google Scholar] [CrossRef]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Lee, Y.S.; Lee, M.; Lee, H.J.; Lee, B.W.; Lee, H.J.; Lee, K.E.; Jung, M.K.; et al. Radioresistant cancer cells can be conditioned to enter senescence by mTOR inhibition. Cancer Res. 2013, 73, 4267–4277. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Xu, M.; Hua, R.; Tan, C.; Zhang, J.; Gong, Y.; Wu, Z.; Weng, W.; Sheng, W.; Guo, W. The polycomb group protein EZH2 induces epithelial-mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J. Hematol. Oncol. 2018, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Hung, M.C. The role of EZH2 in tumour progression. Br. J. Cancer 2012, 106, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, P.W.; Slape, C.I.; Farnsworth, R.H.; Atapattu, L.; Scott, A.M.; Vail, M.E. EphA3 biology and cancer. Growth Factors 2014, 32, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, W.; Yao, C.; Zhang, S.; Liang, L.; Han, M.; Ren, J.; Qi, X.; Zhang, X.; Wang, S.; et al. Radiation-resistant cancer stem-like cell properties are regulated by PTEN through the activity of nuclear β-catenin in nasopharyngeal carcinoma. Oncotarget 2017, 8, 74661–74672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.L.; Wang, C.Y.; Fu, J.; Yang, X.J.; Sun, Y.; Shao, Y.H.; Zhang, L.H.; Yang, X.M.; Zhang, X.L.; Lin, J. PTEN expression in U251 glioma cells enhances their sensitivity to ionizing radiation by suppressing DNA repair capacity. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10453–10458. [Google Scholar] [CrossRef]
- Peng, J.; Lv, Y.; Wu, C. Radiation-resistance increased by overexpression of microRNA-21 and inhibition of its target PTEN in esophageal squamous cell carcinoma. J. Int. Med. Res. 2020, 48, 300060519882543. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Licona, A.; Pérez-Añorve, I.X.; Flores-Fortis, M.; Moral-Hernández, O.D.; González-de la Rosa, C.H.; Suárez-Sánchez, R.; Chávez-Saldaña, M.; Aréchaga-Ocampo, E. Deciphering the epigenetic network in cancer radioresistance. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2021, 159, 48–59. [Google Scholar] [CrossRef]
- Peng, Q.; Weng, K.; Li, S.; Xu, R.; Wang, Y.; Wu, Y. A Perspective of Epigenetic Regulation in Radiotherapy. Front. Cell Dev. Biol. 2021, 9, 624312. [Google Scholar] [CrossRef]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.; Rizzo, S.; van der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar] [CrossRef] [Green Version]
- Bhatla, T.; Wang, J.; Morrison, D.J.; Raetz, E.A.; Burke, M.J.; Brown, P.; Carroll, W.L. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood 2012, 119, 5201–5210. [Google Scholar] [CrossRef] [Green Version]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.K.; Li, Y.; Pandit, H.; Li, S.; Pulliam, Z.; Zheng, Q.; Yu, Y.; Martin, R.C.G. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell. Immunol. 2019, 336, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, G.K.; Mutton, L.N.; Khalili, M.; McMullin, R.P.; Hicks, J.L.; Bianchi-Frias, D.; Horn, L.A.; Kulac, I.; Moubarek, M.S.; Nelson, P.S.; et al. Combined MYC Activation and Pten Loss Are Sufficient to Create Genomic Instability and Lethal Metastatic Prostate Cancer. Cancer Res. 2016, 76, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Surgery Specimens (N = 59) | Recurred Cancer Specimens (N = 45) | p Value | |
|---|---|---|---|
| EPHA3 | |||
| Negative | 28 | 11 | 0.016 |
| Positive | 31 | 34 | |
| c-Myc | |||
| Negative | 30 | 23 | 0.040 |
| Positive | 22 | 29 | |
| PTEN | |||
| Negative | 23 | 30 | 0.006 |
| Positive | 32 | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-H.; Kang, B.-C.; Seong, D.; Lee, W.-H.; An, J.-H.; Je, H.-U.; Cha, H.-J.; Chang, H.-W.; Kim, S.-Y.; Kim, S.-W.; et al. EPHA3 Contributes to Epigenetic Suppression of PTEN in Radioresistant Head and Neck Cancer. Biomolecules 2021, 11, 599. https://doi.org/10.3390/biom11040599
Kim S-H, Kang B-C, Seong D, Lee W-H, An J-H, Je H-U, Cha H-J, Chang H-W, Kim S-Y, Kim S-W, et al. EPHA3 Contributes to Epigenetic Suppression of PTEN in Radioresistant Head and Neck Cancer. Biomolecules. 2021; 11(4):599. https://doi.org/10.3390/biom11040599
Chicago/Turabian StyleKim, Song-Hee, Byung-Chul Kang, Daseul Seong, Won-Hyeok Lee, Jae-Hee An, Hyoung-Uk Je, Hee-Jeong Cha, Hyo-Won Chang, Sang-Yoon Kim, Seong-Who Kim, and et al. 2021. "EPHA3 Contributes to Epigenetic Suppression of PTEN in Radioresistant Head and Neck Cancer" Biomolecules 11, no. 4: 599. https://doi.org/10.3390/biom11040599
APA StyleKim, S. -H., Kang, B. -C., Seong, D., Lee, W. -H., An, J. -H., Je, H. -U., Cha, H. -J., Chang, H. -W., Kim, S. -Y., Kim, S. -W., & Han, M. -W. (2021). EPHA3 Contributes to Epigenetic Suppression of PTEN in Radioresistant Head and Neck Cancer. Biomolecules, 11(4), 599. https://doi.org/10.3390/biom11040599