
Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Analysis
2.2. Virtual Screening
2.3. Cell Lines and Cell Culture
2.4. Flow Cytometry Analysis
2.5. Blocking Assays
2.6. Microscale Thermophoresis (MST)
2.7. Phagocytosis Assays
2.8. Tumor Models and Ex Vivo Assays
2.9. Statistical Analysis
3. Results
3.1. CD47 and PVR Are Over-Expressed in Tumor Tissues and Cell Lines
3.2. Discovery of Small Molecule Inhibitors Targeting CD47/ SIRPα and TIGIT/PVR Pathways by Virtual Screening
3.3. Screening and Validation of the Candidate Small Molecule Inhibitors by Blocking Assay
3.4. The Binding Activity and Model of Azelnidipine to SIRPα and PVR
3.5. Azelnidipine Could Enhance the Macrophages-Mediated Phagocytosis of Tumor Cells In Vitro
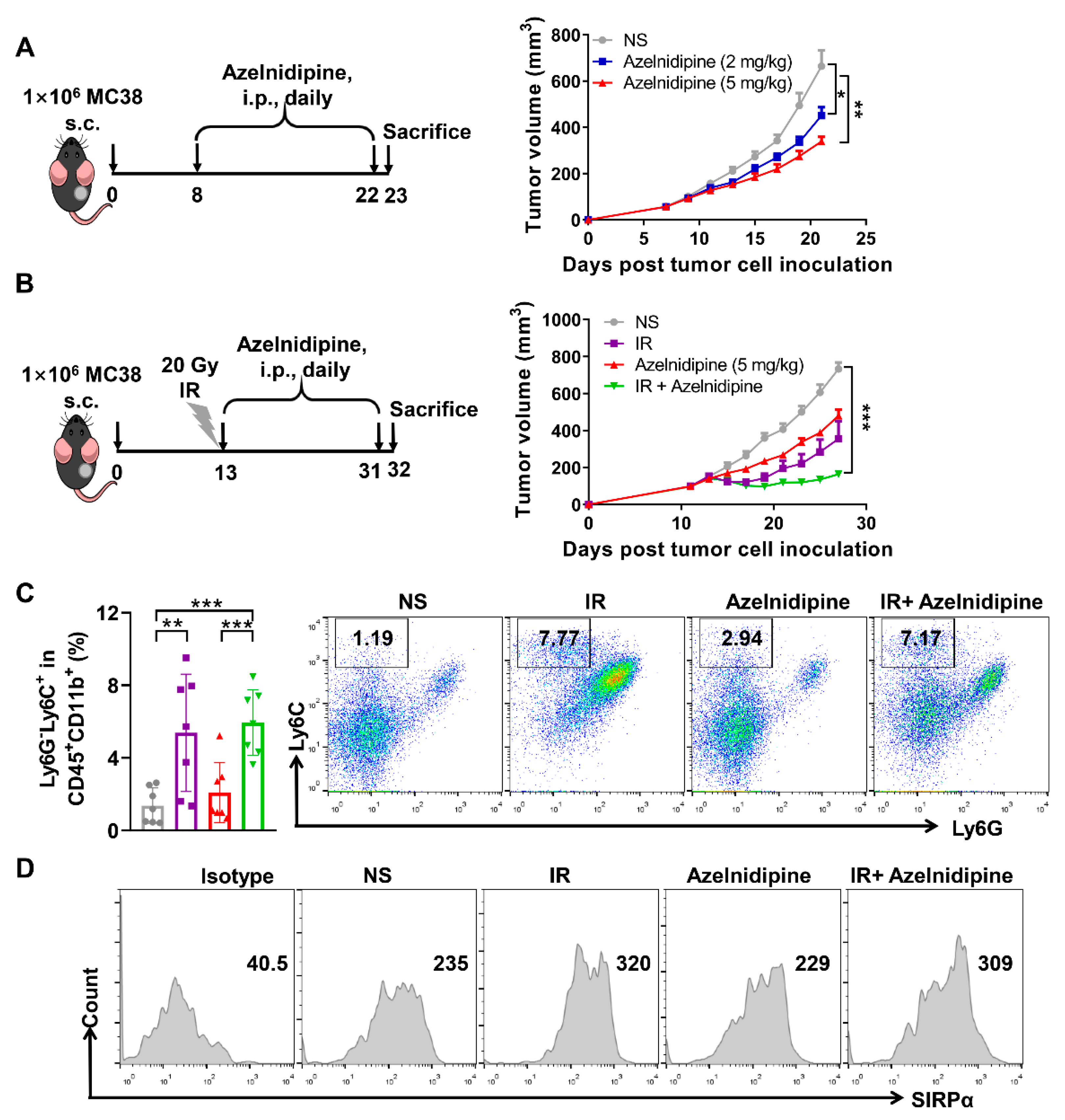
3.6. Azelnidipine Could Inhibit MC38 Tumor Growth Combined with Radiotherapy
3.7. Azelnidipine Could Significantly Inhibit the Tumor Growth and Elicit Anti-Tumor T Cell Immune Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Zhou, X.; Zuo, C.; Li, W.; Shi, W.; Zhou, X.; Wang, H.; Chen, S.; Du, J.; Chen, G.; Zhai, W.; et al. A novel D peptide identified by mirror-image phage display blocks TIGIT/PVR for cancer immunotherapy. Angew. Chem. (Int. Ed. Engl.) 2020. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Bowers, J.R.; Readler, J.M.; Sharma, P.; Excoffon, K. Poliovirus receptor: More than a simple viral receptor. Virus Res. 2017, 242, 1–6. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Madore, J.; Li, X.Y.; Smyth, M.J. Tumor intrinsic and extrinsic immune functions of CD155. Semin. Cancer Biol. 2020, 65, 189–196. [Google Scholar] [CrossRef]
- Stengel, K.F.; Harden-Bowles, K.; Yu, X.; Rouge, L.; Yin, J.P.; Comps-Agrar, L.; Wiesmann, C.; Bazan, J.F.; Eaton, D.L.; Grogan, J.L. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc. Natl. Acad. Sci. USA 2012, 109, 5399–5404. [Google Scholar] [CrossRef] [Green Version]
- Deuss, F.A.; Watson, G.M.; Fu, Z.; Rossjohn, J.; Berry, R. Structural basis for CD96 immune receptor recognition of Nectin-like protein-5, CD155. Structure 2019, 27, 219–228.e213. [Google Scholar] [CrossRef] [Green Version]
- Inozume, T.; Yaguchi, T.; Furuta, J.; Harada, K.; Kawakami, Y.; Shimada, S. Melanoma cells control antimelanoma CTL responses via interaction between TIGIT and CD155 in the effector phase. J. Investig. Dermatol. 2016, 136, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Mao, L.; Liu, J.F.; Chen, L.; Yu, G.T.; Yang, L.L.; Wu, H.; Bu, L.L.; Kulkarni, A.B.; Zhang, W.F.; et al. Blockade of TIGIT/CD155 signaling reverses T-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol. Res. 2019, 7, 1700–1713. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, B.; Gao, J.; Xin, N.; Wang, W.; Song, X.; Shao, Y.; Zhao, C. CD155 knockdown promotes apoptosis via AKT/Bcl-2/Bax in colon cancer cells. J. Cell. Mol. Med. 2018, 22, 131–140. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Du, J.; Wang, H.; Chen, C.; Jiao, L.; Cheng, X.; Zhou, X.; Chen, S.; Gou, S.; Zhao, W.; et al. Repositioning liothyronine for cancer immunotherapy by blocking the interaction of immune checkpoint TIGIT/PVR. Cell Commun. Signal. CCS 2020, 18, 142. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef] [Green Version]
- Rothlin, C.V.; Ghosh, S. Lifting the innate immune barriers to antitumor immunity. J. Immuno Ther. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Kuo, T.C.; Chen, A.; Harrabi, O.; Sockolosky, J.T.; Zhang, A.; Sangalang, E.; Doyle, L.V.; Kauder, S.E.; Fontaine, D.; Bollini, S.; et al. Targeting the myeloid checkpoint receptor SIRPalpha potentiates innate and adaptive immune responses to promote anti-tumor activity. J. Hematol. Oncol. 2020, 13, 160. [Google Scholar] [CrossRef]
- Liu, X.; Liu, L.; Ren, Z.; Yang, K.; Xu, H.; Luan, Y.; Fu, K.; Guo, J.; Peng, H.; Zhu, M.; et al. Dual targeting of innate and adaptive checkpoints on tumor cells limits immune evasion. Cell Rep. 2018, 24, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Guo, H.; Xu, J.; Qin, T.; Guo, Q.; Gu, N.; Zhang, D.; Qian, W.; Dai, J.; Hou, S.; et al. Elimination of tumor by CD47/PD-L1 dual-targeting fusion protein that engages innate and adaptive immune responses. mAbs 2018, 10, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Wurth, R.; Thellung, S.; Bajetto, A.; Mazzanti, M.; Florio, T.; Barbieri, F. Drug-repositioning opportunities for cancer therapy: Novel molecular targets for known compounds. Drug Discov. Today 2016, 21, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, A.; Zhao, Z.; Xu, Y.; Lin, J.; Jou, D.; Li, C. Fragment-based drug design and drug repositioning using multiple ligand simultaneous docking (MLSD): Identifying celecoxib and template compounds as novel inhibitors of signal transducer and activator of transcription 3 (STAT3). J. Med. Chem. 2011, 54, 5592–5596. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Yang, W.-H.; Jung, Y.-S.; Cha, J.-h. A new aspect of an old friend: The beneficial effect of metformin on anti-tumor immunity. BMB Rep. 2020, 53, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Luo, M.; Rong, Q.X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.Y.; Fu, L.W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J Immuno Ther. Cancer 2019, 7, 245. [Google Scholar] [CrossRef]
- Hatherley, D.; Harlos, K.; Dunlop, D.C.; Stuart, D.I.; Barclay, A.N. The structure of the macrophage signal regulatory protein alpha (SIRPalpha) inhibitory receptor reveals a binding face reminiscent of that used by T cell receptors. J. Biol. Chem. 2007, 282, 14567–14575. [Google Scholar] [CrossRef] [Green Version]
- Zhai, W.; Zhou, X.; Du, J.; Gao, Y. In vitro assay for the development of small molecule inhibitors targeting PD-1/PD-L1. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar] [CrossRef]
- Zhai, W.; Zhou, X.; Wang, H.; Li, W.; Chen, G.; Sui, X.; Li, G.; Qi, Y.; Gao, Y. A novel cyclic peptide targeting LAG-3 for cancer immunotherapy by activating antigen-specific CD8(+) T cell responses. Acta Pharm. Sin. B 2020, 10, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPalpha immune checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef]
- Burugu, S.; Dancsok, A.R.; Nielsen, T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Candas-Green, D.; Xie, B.; Huang, J.; Fan, M.; Wang, A.; Menaa, C.; Zhang, Y.; Zhang, L.; Jing, D.; Azghadi, S.; et al. Dual blockade of CD47 and HER2 eliminates radioresistant breast cancer cells. Nat. Commun. 2020, 11, 4591. [Google Scholar] [CrossRef]
- Pai, S.; Bamodu, O.A.; Lin, Y.K.; Lin, C.S.; Chu, P.Y.; Chien, M.H.; Wang, L.S.; Hsiao, M.; Yeh, C.T.; Tsai, J.T. CD47-SIRPalpha signaling induces epithelial-mesenchymal transition and cancer stemness and links to a poor prognosis in patients with oral squamous cell carcinoma. Cells 2019, 8, 1658. [Google Scholar] [CrossRef] [Green Version]
- Kerr, W.G.; Chisholm, J.D. The next generation of immunotherapy for cancer: Small molecules could make big waves. J. Immunol. 2019, 202, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Huck, B.R.; Kötzner, L.; Urbahns, K. Small molecules drive big improvements in immuno-oncology therapies. Angew. Chem. (Int. Ed. Engl.) 2018, 57, 4412–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatherley, D.; Graham, S.C.; Turner, J.; Harlos, K.; Stuart, D.I.; Barclay, A.N. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol. Cell 2008, 31, 266–277. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Zhou, X.; Du, J.; Zhou, X.; Niu, X.; Li, W.; Chen, C.; Lv, S.; Wu, A.; Gou, S.; Sun, Y.; et al. Computer-aided design of PVR mutants with enhanced binding affinity to TIGIT. Cell Commun. Signal. CCS 2021, 19, 12. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, Y.; Zhou, X.; Chen, C.; Jiao, L.; Li, W.; Gou, S.; Li, Y.; Du, J.; Chen, G.; et al. CD47/SIRPα blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. J. Immuno Ther. Cancer 2020, 8, e000905. [Google Scholar] [CrossRef]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.X.; Xu, M.M. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazama, D.; Yin, Y.; Murata, Y.; Matsuda, M.; Okamoto, T.; Tanaka, D.; Terasaka, N.; Zhao, J.; Sakamoto, M.; Kakuchi, Y.; et al. Macrocyclic peptide-mediated blockade of the CD47-SIRPalpha interaction as a potential cancer immunotherapy. Cell Chem. Biol. 2020, 27, 1181–1191.e1187. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Banas, H.; Casas-Aviles, I.; Duran, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: Novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef] [Green Version]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Birx, D.L.; Berger, M.; Fleisher, T.A. The interference of T cell activation by calcium channel blocking agents. J. Immunol. 1984, 133, 2904–2909. [Google Scholar]
- Wright, B.; Zeidman, I.; Greig, R.; Poste, G. Inhibition of macrophage activation by calcium channel blockers and calmodulin antagonists. Cell. Immunol. 1985, 95, 46–53. [Google Scholar] [CrossRef]
- Liu, W.; Matsumori, A. Calcium channel blockers and modulation of innate immunity. Curr. Opin. Infect. Dis. 2011, 24, 254–258. [Google Scholar] [CrossRef]
- Matsumori, A.; Nishio, R.; Nose, Y. Calcium channel blockers differentially modulate cytokine production by peripheral blood mononuclear cells. Circ. J. Off. J. Jpn. Circ. Soc. 2010, 74, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Komoda, H.; Shiraki, A.; Oyama, J.I.; Nishikido, T.; Node, K. Azelnidipine inhibits the differentiation and activation of THP-1 macrophages through the L-type calcium channel. J. Atheroscler. Thromb. 2018, 25, 690–697. [Google Scholar] [CrossRef] [Green Version]
- Miura, R.; Nakamura, K.; Miura, D.; Miura, A.; Kajiya, M.; Hisamatsu, K.; Nagase, S.; Morita, H.; Kusano, K.F.; Matsubara, H.; et al. Cytokine reducing effect of azelnidipine in human peripheral blood mononuclear cells. Biol. Pharm. Bull. 2010, 33, 1148–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Fuse, I.; Narita, M.; Takahashi, M.; Aizawa, Y. Combination use of immune complexes and a Ca2(+) channel blocker azelnidipine enhances interleukin-12 p40 secretion without T helper type 17 cytokine secretion in human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2009, 156, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Chiu-Hung, C.; Show-Mei, C.; Tzong-Der, W.; Nai-Wan, H. Use of Azelnidipine in Preaparing Medicinal Composition for Treating Cancers. Patent EP3222278(A4), 22 October 2015. [Google Scholar]
- Chiu-Hung, C.; Show-Mei, C.; Tzong-Der, W.; Nai-Wan, H. New Indication of Azelnidipine Pharmaceutical Composition for Treating Cancer. Patent US2017312260(A1), 22 October 2015. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Jiao, L.; Qian, Y.; Dong, Q.; Sun, Y.; Zheng, W.V.; Zhao, W.; Zhai, W.; Qiu, L.; Wu, Y.; et al. Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy. Biomolecules 2021, 11, 706. https://doi.org/10.3390/biom11050706
Zhou X, Jiao L, Qian Y, Dong Q, Sun Y, Zheng WV, Zhao W, Zhai W, Qiu L, Wu Y, et al. Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy. Biomolecules. 2021; 11(5):706. https://doi.org/10.3390/biom11050706
Chicago/Turabian StyleZhou, Xiuman, Ling Jiao, Yuzhen Qian, Qingyu Dong, Yixuan Sun, Wei V. Zheng, Wenshan Zhao, Wenjie Zhai, Lu Qiu, Yahong Wu, and et al. 2021. "Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy" Biomolecules 11, no. 5: 706. https://doi.org/10.3390/biom11050706
APA StyleZhou, X., Jiao, L., Qian, Y., Dong, Q., Sun, Y., Zheng, W. V., Zhao, W., Zhai, W., Qiu, L., Wu, Y., Wang, H., Gao, Y., & Chen, J. (2021). Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy. Biomolecules, 11(5), 706. https://doi.org/10.3390/biom11050706