
Circulating Biomarkers Reflecting Destabilization Mechanisms of Coronary Artery Plaques: Are We Looking for the Impossible?

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Pathophysiology of Vulnerable Plaques
3. Biomarkers of Vulnerable Plaques
3.1. Inflammation-Based Biomarkers
3.2. Lipid-Based Biomarkers
3.3. Non-Coding RNAs
3.3.1. MiRNAs
3.3.2. Long Non-Coding RNA (lncRNA)
4. Role of Invasive Imaging Methods in Plaque Pathobiology Assessment
5. Will There Be a Biomarker for Unstable Plaque in the Future?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanchis-Gomar, F.; Perez-Quilis, C.; Leischik, R.; Lucia, A. Epidemiology of coronary heart disease and acute coronary syndrome. Ann. Transl. Med. 2016, 4, 256. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.J.O.; Floriano, J.F.; Nicastro, L.; Emanueli, C.; Catapano, F. Novel Applications of Mesenchymal Stem Cell-derived Exosomes for Myocardial Infarction Therapeutics. Biomolecules 2020, 10, 707. [Google Scholar] [CrossRef]
- Ortega-Rodríguez, A.C.; Marín-Jáuregui, L.S.; Martínez-Shio, E.; Hernández Castro, B.; González-Amaro, R.; Escobedo-Uribe, C.D.; Monsiváis-Urenda, A.E. Altered NK cell receptor repertoire and function of natural killer cells in patients with acute myocardial infarction: A three-month follow-up study. Immunobiology 2020, 225, 151909. [Google Scholar] [CrossRef]
- Naghavi, M.; Libby, P.; Falk, E.; Casscells, S.W.; Litovsky, S.; Rumberger, J.; Badimon, J.J.; Stefanadis, C.; Moreno, P.; Pasterkamp, G.; et al. From vulnerable plaque to vulnerable patient: A call for new definitions and risk assessment strategies: Part II. Circulation 2003, 108, 1772–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodgie, F.D.; Virmani, R.; Burke, A.P.; Farb, A.; Weber, D.K.; Kutys, R.; Finn, A.V.; Gold, H.K. Pathologic assessment of the vulnerable human coronary plaque. Heart 2004, 90, 1385–1391. [Google Scholar] [CrossRef] [Green Version]
- van Lammeren, G.; Moll, F.; Borst, G.J.; de Kleijn, D.P.; de Vries, J.P.; Pasterkamp, G. Atherosclerotic plaque biomarkers: Beyond the horizon of the vulnerable plaque. Curr. Cardiol. Rev. 2011, 7, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Bourantas, C.V.; Garcia-Garcia, H.M.; Farooq, V.; Maehara, A.; Xu, K.; Généreux, P.; Diletti, R.; Muramatsu, T.; Fahy, M.; Weisz, G.; et al. Clinical and angiographic characteristics of patients likely to have vulnerable plaques: Analysis from the PROSPECT study. JACC Cardiovasc. Imaging 2013, 6, 1263–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Korte, C.L.; Hansen, H.H.; van der Steen, A.F. Vascular ultrasound for atherosclerosis imaging. Interface Focus. 2011, 1, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Ten Kate, G.L.; Sijbrands, E.J.; Valkema, R.; ten Cate, F.J.; Feinstein, S.B.; van der Steen, A.F.; Daemen, M.J.; Schinkel, A.F. Molecular imaging of inflammation and intraplaque vasa vasorum: A step forward to identification of vulnerable plaques? J. Nucl. Cardiol. 2010, 17, 897–912. [Google Scholar] [CrossRef] [Green Version]
- Takaya, N.; Yuan, C.; Chu, B.; Saam, T.; Polissar, N.L.; Jarvik, G.P.; Isaac, C.; McDonough, J.; Natiello, C.; Small, R.; et al. Presence of intraplaque hemorrhage stimulates progression of carotid atherosclerotic plaques: A high-resolution magnetic resonance imaging study. Circulation 2005, 111, 2768–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altaf, N.; Beech, A.; Goode, S.D.; Gladman, J.R.; Moody, A.R.; Auer, D.P.; MacSweeney, S.T. Carotid intraplaque hemorrhage detected by magnetic resonance imaging predicts embolization during carotid endarterectomy. J. Vasc. Surg. 2007, 46, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.J.; Camps-Renom, P.; Giannotti, N.; Marti-Fabregas, J.; Murphy, S.; McNulty, J.; Barry, M.; Barry, P.; Calvet, D.; Coutts, S.B.; et al. Carotid plaque inflammation imaged by (18)f-fluorodeoxyglucose positron emission tomography and risk of early recurrent stroke. Stroke 2019, 50, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Skagen, K.; Johnsrud, K.; Evensen, K.; Scott, H.; Krohg-Sorensen, K.; Reier-Nilsen, F.; Revheim, M.E.; Fjeld, J.G.; Skjelland, M.; Russell, D. Carotid plaque inflammation assessed with (18)f-fdg pet/ct is higher in symptomatic compared with asymptomatic patients. Int. J. Stroke. 2015, 10, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, H.; Bourantas, C.; Bagnall, A.; Mintz, G.S.; Kunadian, V. OCT for the identification of vulnerable plaque in acute coronary syndrome. JACC Cardiovasc. Imaging. 2015, 8, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subban, V.; Raffel, O.C. Optical coherence tomography: Fundamentals and clinical utility. Cardiovasc. Diagn. Ther. 2020, 10, 1389–1414. [Google Scholar] [CrossRef]
- Toutouzas, K.; Karanasos, A.; Tousoulis, D. Optical Coherence Tomography For the Detection of the Vulnerable Plaque. Eur. Cardiol. 2016, 11, 90–95. [Google Scholar] [CrossRef]
- Burke, A.P.; Farb, A.; Malcom, G.T.; Liang, Y.H.; Smialek, J.; Virmani, R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N. Engl. J. Med. 1997, 336, 1276–1282. [Google Scholar] [CrossRef]
- Rutledge, C.A.; Chiba, T.; Redding, K.; Dezfulian, C.; Sims-Lucas, S.; Kaufman, B.A. A novel ultrasound-guided mouse model of sudden cardiac arrest. PLoS ONE 2020, 15, e0237292. [Google Scholar] [CrossRef]
- Puig, N.; Jiménez-Xarrié, E.; Camps-Renom, P.; Benitez, S. Search for Reliable Circulating Biomarkers to Predict Carotid Plaque Vulnerability. Int. J. Mol Sci. 2020, 21, 8236. [Google Scholar] [CrossRef]
- Simionescu, M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 266–274. [Google Scholar] [CrossRef]
- Aguilar-Ballester, M.; Herrero-Cervera, A.; Vinué, Á.; Martínez-Hervás, S.; González-Navarro, H. Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis. Nutrients 2020, 12, 2021. [Google Scholar] [CrossRef]
- Sorci-Thomas, M.G.; Thomas, M.J. Microdomains, Inflammation, and Atherosclerosis. Circ. Res. 2016, 118, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Crea, F.; Libby, P. Acute Coronary Syndromes: The Way Forward from Mechanisms to Precision Treatment. Circulation 2017, 136, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Kumrić, M.; Tičinović Kurir, T.; Borovac, J.A.; Božić, J. The Role of Natural Killer (NK) Cells in Acute Coronary Syndrome: A Comprehensive Review. Biomolecules 2020, 10, 1514. [Google Scholar] [CrossRef]
- Tabas, I.; Tall, A.; Accili, D. The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression. Circ. Res. 2010, 106, 58–67. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Kirichenko, T.V.; Markina, Y.V.; Sukhorukov, V.N.; Khotina, V.A.; Wu, W.K.; Orekhov, A.N. A Novel Insight at Atherogenesis: The Role of Microbiome. Front. Cell Dev. Biol. 2020, 8, 586189. [Google Scholar] [CrossRef] [PubMed]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Atherosclerosis as an inflammatory disease. Curr. Pharm Des. 2012, 18, 4266–4288. [Google Scholar] [CrossRef]
- Virmani, R.; Burke, A.P.; Kolodgie, F.D.; Farb, A. Vulnerable plaque: The pathology of unstable coronary lesions. J. Interv. Cardiol. 2002, 15, 439–446. [Google Scholar] [CrossRef]
- Hafiane, A. Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. J. Cardiovasc. Dev. Dis. 2019, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Borovac, J.A.; Glavas, D.; Susilovic Grabovac, Z.; Supe Domic, D.; D’Amario, D.; Bozic, J. Catestatin in Acutely Decompensated Heart Failure Patients: Insights from the CATSTAT-HF Study. J. Clin. Med. 2019, 8, 1132. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Pasterkamp, G. Requiem for the ‘vulnerable plaque’. Eur. Heart, J. 2015, 36, 2984–2987. [Google Scholar] [CrossRef] [Green Version]
- Stefanadis, C.; Antoniou, C.K.; Tsiachris, D.; Pietri, P. Coronary Atherosclerotic Vulnerable Plaque: Current Perspectives. J. Am. Heart Assoc. 2017, 6, e005543. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.J.; Lin, F.Y.; Lee, S.E.; Andreini, D.; Bax, J.; Cademartiri, F.; Chinnaiyan, K.; Chow, B.J.W.; Conte, E.; Cury, R.C.; et al. Coronary Atherosclerotic Precursors of Acute Coronary Syndromes. J. Am. Coll. Cardiol. 2018, 71, 2511–2522. [Google Scholar] [CrossRef]
- Arbustini, E.; Dal Bello, B.; Morbini, P.; Burke, A.P.; Bocciarelli, M.; Specchia, G.; Virmani, R. Plaque erosion is a major substrate for coronary thrombosis in acute myocardial infarction. Heart 1999, 82, 269–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodgie, F.D.; Burke, A.P.; Farb, A.; Weber, D.K.; Kutys, R.; Wight, T.N.; Virmani, R. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: Insights into plaque erosion. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1642–1648. [Google Scholar] [CrossRef] [Green Version]
- Bilalic, A.; Ticinovic Kurir, T.; Kumric, M.; Borovac, J.A.; Matetic, A.; Supe-Domic, D.; Bozic, J. Circulating Levels of Dephosphorylated-Uncarboxylated Matrix Gla Protein in Patients with Acute Coronary Syndrome. Molecules 2021, 26, 1108. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Scalone, G.; Niccoli, G.; Refaat, H.; Vergallo, R.; Porto, I.; Leone, A.M.; Burzotta, F.; D’Amario, D.; Liuzzo, G.; Fracassi, F.; et al. Not all plaque ruptures are born equal: An optical coherence tomography study. Eur. Heart J. Cardiovasc. Imaging. 2017, 18, 1271–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niccoli, G.; Montone, R.A.; Cataneo, L.; Cosentino, N.; Gramegna, M.; Refaat, H.; Porto, I.; Burzotta, F.; Trani, C.; Leone, A.M.; et al. Morphological-biohumoral correlations in acute coronary syndromes: Pathogenetic implications. Int. J. Cardiol. 2014, 171, 463–466. [Google Scholar] [CrossRef]
- Jia, H.; Abtahian, F.; Aguirre, A.D.; Lee, S.; Chia, S.; Lowe, H.; Kato, K.; Yonetsu, T.; Vergallo, R.; Hu, S.; et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J. Am. Coll. Cardiol. 2013, 62, 1748–1758. [Google Scholar] [CrossRef] [Green Version]
- Borovac, J.A.; D’Amario, D.; Vergallo, R.; Porto, I.; Bisignani, A.; Galli, M.; Annibali, G.; Montone, R.A.; Leone, A.M.; Niccoli, G.; et al. Neoatherosclerosis after drug-eluting stent implantation: A novel clinical and therapeutic challenge. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 105–116. [Google Scholar] [CrossRef]
- Henson, P.M.; Bratton, D.L.; Fadok, V.A. Apoptotic cell removal. Curr. Biol. 2001, 11, R795–R805. [Google Scholar] [CrossRef] [Green Version]
- Kojima, Y.; Volkmer, J.P.; McKenna, K.; Civelek, M.; Lusis, A.J.; Miller, C.L.; Direnzo, D.; Nanda, V.; Ye, J.; Connolly, A.J. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 2016, 536, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 2007, 109, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A., Jr.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Novel lipid mediators and resolution mechanisms in acute inflammation: To resolve or not? Am. J. Pathol. 2010, 177, 1576–1591. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Glass, C.K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 2013, 339, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef] [PubMed]
- Viola, J.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Döring, Y.; Drechsler, M. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ.Res. 2016, 119, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Fredman, G.; Kamaly, N.; Spolitu, S.; Milton, J.; Ghorpade, D.; Chiasson, R.; Kuriakose, G.; Perretti, M.; Farokzhad, O.; Tabas, I. Targeted nanoparticles containing the pro-resolving peptide Ac2-26 protect against advanced atheosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 2015, 7, 275ra20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I. 2016 Russell Ross Memorial Lecture in Vascular Biology: Molecular-Cellular Mechanisms in the Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredman, G.; Ozcan, L.; Spolitu, S.; Hellmann, J.; Spite, M.; Backs, J.; Tabas, I. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 14530–14535. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Serhan, C. Specific lipid mediator signatures of human phagocytes: Microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012. [CrossRef] [Green Version]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.S.; Spite, M.; Fredman, G.; Tabas, I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 6526–6531. [Google Scholar] [CrossRef] [Green Version]
- Peeters, W.; Hellings, W.E.; de Kleijn, D.P.; de Vries, J.P.; Moll, F.L.; Vink, A.; Pasterkamp, G. Carotid atherosclerotic plaques stabilize after stroke: Insights into the natural process of atherosclerotic plaque stabilization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Ylä-Herttuala, S.; Bentzon, J.F.; Daemen, M.; Falk, E.; Garcia-Garcia, H.M.; Herrmann, J.; Hoefer, I.; Jauhiainen, S.; Jukema, J.W.; Krams, R. ESC Working Group of Atherosclerosis and Vascular Biology. Stabilization of atherosclerotic plaques: An update. Eur. Heart J. 2013, 34, 3251–3258. [Google Scholar] [CrossRef] [Green Version]
- Moutachakkir, M.; Lamrani Hanchi, A.; Baraou, A.; Boukhira, A.; Chellak, S. Immunoanalytical characteristics of C-reactive protein and high sensitivity C-reactive protein. Ann Biol. Clin. (Paris). 2017, 75, 225–229, English. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, X.; Cheng, G.; Zhao, C.; Zhang, L.; Hong, Y.; Wan, Q.; He, R.; Wang, Z. Hs-CRP and all-cause, cardiovascular, and cancer mortality risk: A meta-analysis. Atherosclerosis 2017, 259, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Ferreiros, E.R.; Boissonnet, C.P.; Pizarro, R.; Merletti, P.F.; Corrado, G.; Cagide, A.; Bazzino, O.O. Independent prognostic value of elevated C-reactive protein in unstable angina. Circulation 1999, 100, 1958–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagrand, W.K.; Visser, C.A.; Hermens, W.T.; Niessen, H.W.; Verheugt, F.W.; Wolbink, G.J.; Hack, C.E. C-reactive protein as a cardiovascular risk factor: More than an epiphenomenon? Circulation 1999, 100, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, T.; Hatakeyama, K.; Imamura, T.; Date, H.; Shibata, Y.; Hikichi, Y.; Asada, Y.; Eto, T. Involvement of C-reactive protein obtained by directional coronary atherectomy in plaque instability and developing restenosis in patients with stable or unstable angina pectoris. Am. J. Cardiol. 2003, 91, 287–292. [Google Scholar] [CrossRef]
- Inoue, T.; Kato, T.; Uchida, T.; Sakuma, M.; Nakajima, A.; Shibazaki, M.; Imoto, Y.; Saito, M.; Hashimoto, S.; Hikichi, Y.; et al. Local release of C-reactive protein from vulnerable plaque or coronary arterial wall injured by stenting. J. Am. Coll. Cardiol. 2005, 46, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Norja, S.; Nuutila, L.; Karhunen, P.J.; Goebeler, S. C-reactive protein in vulnerable coronary plaques. J. Clin. Pathol. 2007, 60, 545–548. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, P.; Golino, P.; Calabrò, P.; Calì, G.; Ragni, M.; De Rosa, S.; Cimmino, G.; Pacileo, M.; De Palma, R.; Forte, L.; et al. C-reactive protein induces tissue factor expression and promotes smooth muscle and endothelial cell proliferation. Cardiovasc. Res. 2005, 68, 47–55. [Google Scholar] [CrossRef] [Green Version]
- S Hackam, D.G.; Shumak, S.L. C-reactive protein for the prediction of cardiovascular risk: Ready for prime-time? CMAJ. 2004, 170, 1563–1565. [Google Scholar] [CrossRef] [Green Version]
- Sabatine, M.S.; Morrow, D.A.; Jablonski, K.A.; Rice, M.M.; Warnica, J.W.; Domanski, M.J.; Hsia, J.; Gersh, B.J.; Rifai, N.; Ridker, P.M.; et al. Prognostic significance of the Centers for Disease Control/American Heart Association high-sensitivity C-reactive protein cut points for cardiovascular and other outcomes in patients with stable coronary artery disease. Circulation 2007, 115, 1528–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.L. Matrix metalloproteinases: Influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev. Cardiovasc. Ther. 2007, 5, 265–282. [Google Scholar] [CrossRef]
- Xu, X.P.; Meisel, S.R.; Ong, J.M.; Kaul, S.; Cercek, B.; Rajavashisth, T.B.; Sharifi, B.; Shah, P.K. Oxidized low-density lipoprotein regulates matrix metallo- proteinase-9 and its tissue inhibitor in human monocyte-derived macrophages. Circulation 1999, 99, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Kalela, A.; Koivu, T.A.; Sisto, T.; Kanervisto, J.; Hoyhtya, M.; Sillanaukee, P.; Lehtimaki, T.; Nikkari, S.T. Serum matrix met-alloproteinase-9 concentration in angiographically assessed coronary artery disease. Scand J. Clin Lab Invest 2002, 62, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.Y.; Zhang, B.; Yan, Y.H.; Gao, S.S.; Liu, J.J.; Xu, L.; Hui, P.J. Specific matrix metalloproteinases and calcification factors are associated with the vulnerability of human carotid plaque. Exp. Ther. Med. 2018, 16, 2071–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, S.R.; Willeit, K.; Didangelos, A.; Matic, L.P.; Skroblin, P.; Barallobre-Barreiro, J.; Lengquist, M.; Rungger, G.; Kapustin, A.; Kedenko, L.; et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J. Clin. Investig. 2017, 127, 1546–1560. [Google Scholar] [CrossRef] [PubMed]
- Sukhova, G.K.; Schonbeck, U.; Rabkin, E.; Schoen, F.J.; Poole, R.; Billinghurst, R.C.; Libby, P. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation 1999, 99, 2503–2509. [Google Scholar] [CrossRef]
- Sluijter, J.P.G.; Pulskens, W.P.C.; Schoneveld, A.H.; Velema, E.; Strijder, C.F.; Moll, F.; de Vries, J.P.; Verheijen, J.; Hanemaaijer, R.; de Kleijn, D.P.; et al. Matrix metalloproteinase 2 is associated with stable and matrix metalloproteinases 8 and 9 with vulnerable carotid atherosclerotic lesions-a study in hu-manendarterectomy specimen pointing to a role for different extracellular matrix metalloproteinase inducer glycosylation forms. Stroke 2006, 37, 235–239. [Google Scholar]
- Kai, H.; Ikeda, H.; Yasukawa, H.; Kai, M.; Seki, Y.; Kuwahara, F.; Ueno, T.; Sugi, K.; Imaizumi, T. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J. Am. Coll. Cardiol. 1998, 32, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, D.; Shimada, K.; Tanaka, A.; Kusuyama, T.; Yamashita, H.; Ehara, S.; Nakamura, Y.; Kawarabayashi, T.; Iida, H.; Yoshiyama, M.; et al. Comparison of Levels of Serum Matrix Metalloproteinase-9 in Patients With Acute Myocardial Infarction Versus Unstable Angina Pectoris Versus Stable Angina Pectoris. Am. J. Cardiol. 2006, 97, 175–180. [Google Scholar] [CrossRef]
- Ferroni, P.; Basili, S.; Martini, F.; Cardarello, C.M.; Ceci, F.; Di Franco, M.; Bertazzoni, G.; Gazzaniga, P.P.; Alessandri, C. Serum Metalloproteinase 9 Levels in Patients with Coronary Artery Disease: A Novel Marker of Inflammation. J. Investig. Med. 2003, 51, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh-Makoui, R.; Razi, B.; Aslani, S.; Imani, D.; Tabaee, S.S. The association between Matrix Metallo-proteinases-9 (MMP-9) gene family polymorphisms and risk of Coronary Artery Disease (CAD): A systematic review and meta-analysis. BMC Cardiovasc. Disord. 2020, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-J.; Chae, I.-H.; Park, K.-W.; Ju, J.-R.; Oh, S.; Lee, M.-M.; Park, Y.-B. Functional polymorphism in the promoter region of the gelatinase B gene in relation to coronary artery disease and restenosis after percutaneous coronary intervention. J. Hum. Genet. 2002, 47, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wang, Z.; Chang, L.; Chen, G. Association of MMP-9 polymorphisms with diabetic nephropathy risk. Medicine 2020, 99, e22278. [Google Scholar] [CrossRef] [PubMed]
- Küchler, E.C.; Barreiros, D.; Da Silva, R.O.; De Abreu, J.G.B.; Teixeira, E.C.; Da Silva, R.A.B.; Da Silva, L.A.B.; Filho, P.N.; Romano, F.L.; Granjeiro, J.M.; et al. Genetic Polymorphism in MMP9 May Be Associated With Anterior Open Bite in Children. Braz. Dent. J. 2017, 28, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Blankenberg, S.; Rupprecht, H.J.; Poirier, O.; Bickel, C.; Smieja, M.; Hafner, G.; Meyer, J.; Cambien, F.; Tiret, L. Plasma Concentrations and Genetic Variation of Matrix Metalloproteinase 9 and Prognosis of Patients With Cardiovascular Disease. Circulation 2003, 107, 1579–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-X.; Lü, S.-Z.; Zhang, W.-J.; Song, X.-T.; Chen, H.; Zhang, L.-J. Comparision of high sensitivity C-reactive protein and matrix metalloproteinase 9 in patients with unstable angina between with and without significant coronary artery plaques. Chin. Med. J. 2011, 124, 1657–1661. [Google Scholar] [PubMed]
- Sivalingam, Z.; Larsen, S.B.; Grove, E.L.; Hvas, A.-M.; Kristensen, S.D.; Magnusson, N.E. Neutrophil gelatinase-associated lipocalin as a risk marker in cardiovascular disease. Clin. Chem. Lab. Med. 2017, 56, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Eilenberg, W.; Stojkovic, S.; Kaider, A.; Piechota-Polanczyk, A.; Nanobachvili, J.; Domenig, C.M.; Wojta, J.; Huk, I.; Demyanets, S.; Neumayer, C. Neutrophil Gelatinase Associated Lipocalin (NGAL) for Identification of Unstable Plaques in Patients with Asymptomatic Carotid Stenosis. Eur. J. Vasc. Endovasc. Surg. 2019, 57, 768–777. [Google Scholar] [CrossRef]
- Sahinarslan, A.; Kocaman, S.A.; Bas, D.; Akyel, A.; Ercin, U.; Zengin, O.; Timurkaynak, T. Plasma neutrophil gelatinase-associated lipocalin levels in acute myocardial infarction and stable coronary artery disease. Coron. Artery Dis. 2011, 22, 333–338. [Google Scholar] [CrossRef]
- Choi, K.M.; Lee, J.S.; Kim, E.J.; Baik, S.H.; Seo, H.S.; Choi, D.S.; Oh, D.J.; Park, C.G. Implication of lipocalin-2 and visfatin levels in patients with coronary heart disease. Eur. J. Endocrinol. 2008, 158, 203–207. [Google Scholar] [CrossRef]
- Giurgea, G.-A.; Zlabinger, K.; Gugerell, A.; Lukovic, D.; Syeda, B.; Mandic, L.; Pavo, N.; Mester-Tonczar, J.; Traxler-Weidenauer, D.; Spannbauer, A.; et al. Multimarker Approach to Identify Patients with Coronary Artery Disease at High Risk for Subsequent Cardiac Adverse Events: The Multi-Biomarker Study. Biomolecules 2020, 10, 909. [Google Scholar] [CrossRef]
- Lindberg, S.; Pedersen, S.H.; Mogelvang, R.; Jensen, J.S.; Flyvbjerg, A.; Galatius, S.; Magnusson, N.E. Prognostic Utility of Neutrophil Gelatinase-Associated Lipocalin in Predicting Mortality and Cardiovascular Events in Patients With ST-Segment Elevation Myocardial Infarction Treated With Primary Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2012, 60, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zykov, M.Z.; Kashtalap, V.K.; Bykova, I.B.; Hryachkova, O.H.; Kalaeva, V.K.; Shafranskaya, K.S.; Karetnikova, V.K.; Barbarash, O.B. Institute for Complex Problems of Сardiovascular Disease Clinical and Prognostic Value of Serum Neutrophil Gelatinase-Associated Lipocalin in Patients With ST-Segment Elevation Myocardial Infarction. Kardiologiia 2016, 56, 24–29. [Google Scholar] [CrossRef]
- Woitas, R.P.; Scharnagl, H.; Kleber, M.E.; Delgado, G.E.; Grammer, T.B.; Pichler, M.; Krämer, B.K.; März, W.; Stojakovic, T. Neutrophil gelatinase-associated lipocalin levels are U-shaped in the Ludwigshafen Risk and Cardiovascular Health (LURIC) study—Impact for mortality. PLoS ONE 2017, 12, e0171574. [Google Scholar] [CrossRef] [Green Version]
- Kumrić, M.; Borovac, J.; Kurir, T.; Božić, J. Clinical Implications of Uric Acid in Heart Failure: A Comprehensive Review. Life 2021, 11, 53. [Google Scholar] [CrossRef]
- Murase, T.; Kume, N.; Kataoka, H.; Minami, M.; Sawamura, T.; Masaki, T.; Kita, T. Identification of Soluble Forms of Lectin-Like Oxidized LDL Receptor-1. Arter. Thromb. Vasc. Biol. 2000, 20, 715–720. [Google Scholar] [CrossRef] [Green Version]
- Kume, N.; Kita, T. New scavenger receptors and their functions in atherogenesis. Curr. Atheroscler. Rep. 2002, 4, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Morimoto, M.; Hayashida, K.; Hashimoto, N.; Kita, T. Oxidized LDL Modulates Bax/Bcl-2 Through the Lectinlike Ox-LDL Receptor-1 in Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2001, 21, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Kume, N.; Kita, T. Apoptosis of vascular cells by oxidized LDL: Involvement of caspases and LOX-1, and its implication in atherosclerotic plaque rupture. Circ. Res. 2004, 94, 269–270. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, L.; Chen, H.; Sawamura, T.; Ranganathan, S.; Mehta, J.L. LOX-1 Mediates Oxidized Low-Density Lipoprotein-Induced Expression of Matrix Metalloproteinases in Human Coronary Artery Endothelial Cells. Circulation 2003, 107, 612–617. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, K.; Kume, N.; Murase, T.; Minami, M.; Nakagawa, D.; Inada, T.; Minami, M.; Nakagawa, D.; Inada, T.; Tanaka, M.; et al. Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome: A novel marker for early diagnosis. Circulation 2005, 112, 812–818. [Google Scholar] [CrossRef]
- Ueda, A.; Kume, N.; Hayashida, K.; Inui-Hayashida, A.; Asai, M.; Kita, T.; Kominami, G. ELISA for Soluble Form of Lectin-Like Oxidized LDL Receptor-1, A Novel Marker of Acute Coronary Syndrome. Clin. Chem. 2006, 52, 1210–1211. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Takano, M.; Hata, N.; Kume, N.; Yamamoto, M.; Yokoyama, S.; Shinada, T.; Tomita, K.; Shirakabe, A.; Otsuka, T.; et al. Soluble lectin-like oxidized LDL receptor-1 (sLOX-1) as a valuable diagnostic marker for rupture of thin-cap fibroatheroma: Verification by optical coherence tomography. Int. J. Cardiol. 2013, 168, 3217–3223. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Hata, N.; Kume, N.; Shinada, T.; Tomita, K.; Shirakabe, A.; Kitamura, M.; Nozaki, A.; Inami, T.; Seino, Y.; et al. Soluble Lectin-Like Oxidized LDL Receptor-1 and High-Sensitivity Troponin T as Diagnostic Biomarkers for Acute Coronary Syndrome–Improved Values With Combination Usage in Emergency Rooms–. Circ. J. 2011, 75, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Li, Y.; Chen, S.; Yang, X.; Liu, F.; Li, Y.; Li, J.; Cao, J.; Liu, X.; Chen, J.; et al. Association of Lipids With Ischemic and Hemorrhagic Stroke. Stroke 2019, 50, 3376–3384. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yue, Y.; Luo, Y.; Wang, W.; Cao, Y.; Fang, Q. Clinical Utility of the Inflammatory Factors Combined With Lipid Markers in the Diagnostic and Prognostic Assessment of Ischemic Stroke: Based on Logistic Regression Models. J. Stroke Cerebrovasc. Dis. 2020, 29, 104653. [Google Scholar] [CrossRef]
- Claessen, B.E.; Guedeney, P.; Gibson, C.M.; Angiolillo, D.J.; Cao, D.; Lepor, N.; Mehran, R. Lipid Management in Patients Presenting With Acute Coronary Syndromes: A Review. J. Am. Hear. Assoc. 2020, 9, e018897. [Google Scholar] [CrossRef]
- Ke, L.-Y.; Law, S.H.; Mishra, V.K.; Parveen, F.; Chan, H.-C.; Lu, Y.-H.; Chu, C.-S. Molecular and Cellular Mechanisms of Electronegative Lipoproteins in Cardiovascular Diseases. Biomedicines 2020, 8, 550. [Google Scholar] [CrossRef]
- Lehti, S.; Nguyen, S.D.; Belevich, I.; Vihinen, H.; Heikkilä, H.M.; Soliymani, R.; Käkelä, R.; Saksi, J.; Jauhiainen, M.; Grabowski, G.A.; et al. Extracellular Lipids Accumulate in Human Carotid Arteries as Distinct Three-Dimensional Structures and Have Proinflammatory Properties. Am. J. Pathol. 2018, 188, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Hartley, A.; Haskard, D.; Khamis, R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis – Novel insights and future directions in diagnosis and therapy. Trends Cardiovasc. Med. 2019, 29, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Yang, Y.; Su, Z.; Yue, W.; Hao, H.; Ren, L.; Wang, Y.; Cao, Y.; Wang, Y. Association of Oxidized Low-Density Lipoprotein With Prognosis of Stroke and Stroke Subtypes. Stroke 2017, 48, 91–97. [Google Scholar] [CrossRef]
- Wang, A.; Dai, L.; Zhang, N.; Lin, J.; Chen, G.; Zuo, Y.; Li, H.; Wang, Y.; Meng, X.; Wang, Y. Oxidized low-density lipoprotein (LDL) and LDL cholesterol are associated with outcomes of minor stroke and TIA. Atherosclerosis 2020, 297, 74–80. [Google Scholar] [CrossRef]
- Sigala, F.; Kotsinas, A.; Savari, P.; Filis, K.; Markantonis, S.; Iliodromitis, E.K.; Gorgoulis, V.G.; Andreadou, I. Oxidized LDL in human carotid plaques is related to symptomatic carotid disease and lesion instability. J. Vasc. Surg. 2010, 52, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Fu, B.; He, D.; Zhang, Y.; Liu, J.; Zhang, X. The relationship between oxidized low-density lipoprotein and related ratio and acute cerebral infarction. Medicine 2018, 97, e12642. [Google Scholar] [CrossRef]
- Zhang, Y.-C.; Tang, Y.; Chen, Y.; Huang, X.-H.; Zhang, M.; Chen, J.; Sun, Y.-G.; Li, Y.-G. Oxidized Low-Density Lipoprotein and C-Reactive Protein Have Combined Utility for Better Predicting Prognosis After Acute Coronary Syndrome. Cell Biophys. 2013, 68, 379–385. [Google Scholar] [CrossRef]
- Wilson, P.W.; Ben-Yehuda, O.; McNamara, J.; Massaro, J.; Witztum, J.; Reaven, P.D. Autoantibodies to oxidized LDL and cardiovascular risk: The Framingham Offspring Study. Atherosclerosis 2006, 189, 364–368. [Google Scholar] [CrossRef]
- Holvoet, P.; Kritchevsky, S.B.; Tracy, R.P.; Mertens, A.; Rubin, S.M.; Butler, J.; Goodpaster, B.; Harris, T.B. The metabolic syndrome, circulating oxidized LDL, and risk of myocardial infarction in well-functioning elderly people in the health, aging, and body composition cohort. Diabetes 2004, 53, 1068–1073. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Urbina, A.; Rull, A.; Ordóñez-Llanos, J.; Sanchez-Quesada, J.L. Electronegative LDL: An Active Player in Atherogenesis or a By- Product of Atherosclerosis? Curr. Med. Chem. 2019, 26, 1665–1679. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-S.; Law, S.H.; Lenzen, D.; Tan, Y.-H.; Weng, S.-F.; Ito, E.; Wu, J.-C.; Chen, C.-H.; Chan, H.-C.; Ke, L.-Y. Clinical Significance of Electronegative Low-Density Lipoprotein Cholesterol in Atherothrombosis. Biomedicine 2020, 8, 254. [Google Scholar] [CrossRef]
- Hsu, J.-F.; Chou, T.-C.; Lu, J.; Chen, S.-H.; Chen, F.-Y.; Chen, C.-C.; Chen, J.L.; Elayda, M.; Ballantyne, C.M.; Shayani, S.; et al. Low-Density Lipoprotein Electronegativity Is a Novel Cardiometabolic Risk Factor. PLoS ONE 2014, 9, e107340. [Google Scholar] [CrossRef]
- Chang, C.-T.; Shen, M.-Y.; Lee, A.-S.; Wang, C.-C.; Chen, W.-Y.; Chang, C.-M.; Chang, K.-C.; Stancel, N.; Chen, C.-H. Electronegative low-density lipoprotein increases the risk of ischemic lower-extremity peripheral artery disease in uremia patients on maintenance hemodialysis. Sci. Rep. 2017, 7, 4654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.-S.; Yang, T.-C.; Chang, P.-Y.; Chang, S.-F.; Ho, S.-L.; Chen, H.-L.; Lu, S.-C. Electronegative LDL is linked to high-fat, high-cholesterol diet–induced nonalcoholic steatohepatitis in hamsters. J. Nutr. Biochem. 2016, 30, 44–52. [Google Scholar] [CrossRef]
- Estruch, M.; Miñambres, I.; Sanchez-Quesada, J.L.; Soler, M.; Pérez, A.; Ordoñez-Llanos, J.; Benitez, S. Increased inflammatory effect of electronegative LDL and decreased protection by HDL in type 2 diabetic patients. Atherosclerosis 2017, 265, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, J.H.; Burns, A.R.; Tang, D.; Walterscheid, J.P.; Suzuki, S.; Yang, C.Y.; Sawamura, T.; Chen, C.H. Mediation of electronegative low-density lipoprotein signaling by LOX-1: A possible mechanism of endothelial apoptosis. Circ. Res. 2009, 104, 619–627. [Google Scholar] [CrossRef]
- Yang, T.-C.; Chang, P.-Y.; Lu, S.-C. L5-LDL from ST-elevation myocardial infarction patients induces IL-1β production via LOX-1 and NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Circ. Physiol. 2017, 312, H265–H274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.-Y.; Chen, F.-Y.; Hsu, J.-F.; Fu, R.-H.; Chang, C.-M.; Chang, C.-T.; Liu, C.-H.; Wu, J.-R.; Lee, A.-S.; Chan, H.-C.; et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Bonnefont-Rousselot, D. La Lp-PLA2, marqueur d’inflammation vasculaire et de vulnérabilité de la plaque d’athérosclérose. Annales Pharmaceutiques Françaises 2016, 74, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Burke, A.P.; Skorija, K.S.; Ladich, E.; Kutys, R.; Makuria, A.T.; Virmani, R. Lipoprotein-Associated Phospholipase A 2 Protein Expression in the Natural Progression of Human Coronary Atherosclerosis. Arter. Thromb. Vasc. Biol. 2006, 26, 2523–2529. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, A.; Mannucci, L.; Tamburi, F.; Cardillo, C.; Schinzari, F.; Rovella, V.; Nisticò, S.; Bennardo, L.; Di Daniele, N.; Tesauro, M. Lp-PLA2, a new biomarker of vascular disorders in metabolic diseases. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419827154. [Google Scholar] [CrossRef] [Green Version]
- Nelson, T.L.; Kamineni, A.; Psaty, B.; Cushman, M.; Jenny, N.S.; Hokanson, J.; Furberg, C.; Mukamal, K.J. Lipoprotein-associated phospholipase A2 and future risk of subclinical disease and cardiovascular events in individuals with type 2 diabetes: The Cardiovascular Health Study. Diabetologia 2011, 54, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Sarlon-Bartoli, G.; Boudes, A.; Buffat, C.; Bartoli, M.; Piercecchi-Marti, M.; Sarlon, E.; Arnaud, L.; Bennis, Y.; Thevenin, B.; Squarcioni, C.; et al. Circulating Lipoprotein-associated Phospholipase A2 in High-grade Carotid Stenosis: A New Biomarker for Predicting Unstable Plaque. Eur. J. Vasc. Endovasc. Surg. 2012, 43, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.-L.; Liu, J.-N.; Du, Y.-M.; Yao, G.-H.; Jiang, W.-D.; Wang, X.; Dong, Z.-Q.; Hao, L.; Wang, G.-Y.; Sui, S.-J.; et al. The correlation of human serum Lp-PLA2 and hs-CRP and stability of coronary atherosclerotic plaques. Zhonghua nei ke za zhi 2009, 48, 651–654. [Google Scholar]
- Khuseyinova, N.; Imhof, A.; Rothenbacher, D.; Trischler, G.; Kuelb, S.; Scharnagl, H.; Maerz, W.; Brenner, H.; Koenig, W. Association between Lp-PLA2 and coronary artery disease: Focus on its relationship with lipoproteins and markers of inflammation and hemostasis. Atherosclerosi. 2005, 182, 181–188. [Google Scholar] [CrossRef]
- Thompson, A.; Gao, P.; Orfei, L.; Watson, S.; Di Angelantonio, E.; Kaptoge, S.; Ballantyne, C.; Cannon, C.P.; Criqui, M.; Cushman, M.; et al. Lp-PLA(2) Studies Collaboration. (2010). Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: Collaborative analysis of 32 prospective studies. Lancet 2010, 375, 1536–1544. [Google Scholar] [PubMed] [Green Version]
- Vittos, O.; Toana, B.; Vittos, A.; Moldoveanu, E. Lipoprotein-associated phospholipase A2 (Lp-PLA2): A review of its role and significance as a cardiovascular biomarker. Biomarkers 2012, 17, 289–302. [Google Scholar] [CrossRef]
- Fenning, R.S.; Burgert, M.E.; Hamamdzic, D.; Peyster, E.G.; Mohler, E.R.; Kangovi, S.; Jucker, B.M.; Lenhard, S.C.; Macphee, C.H.; Wilensky, R.L. Atherosclerotic Plaque Inflammation Varies Between Vascular Sites and Correlates With Response to Inhibition of Lipoprotein-Associated Phospholipase A 2. J. Am. Hear. Assoc. 2015, 4, e001477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.T.; Olson, E.N. MicroRNAs in Stress Signaling and Human Disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, H.; Suzuki, H.I. Systems and Synthetic microRNA Biology: From Biogenesis to Disease Pathogenesis. Int. J. Mol. Sci. 2019, 21, 132. [Google Scholar] [CrossRef] [Green Version]
- Cipollone, F.; Felicioni, L.; Sarzani, R.; Ucchino, S.; Spigonardo, F.; Mandolini, C.; Malatesta, S.; Bucci, M.; Mammarella, C.; Santovito, D.; et al. A Unique MicroRNA Signature Associated With Plaque Instability in Humans. Stroke 2011, 42, 2556–2563. [Google Scholar] [CrossRef] [Green Version]
- Soeki, T.; Yamaguchi, K.; Niki, T.; Uematsu, E.; Bando, S.; Matsuura, T.; Ise, T.; Kusunose, K.; Hotchi, J.; Tobiume, T.; et al. Plasma MicroRNA-100 Is Associated With Coronary Plaque Vulnerability. Circ. J. 2015, 79, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Raju, S.; Fish, J.E.; Howe, K.L. MicroRNAs as sentinels and protagonists of carotid artery thromboembolism. Clin. Sci. 2020, 134, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Li, D.Y.; Chernogubova, E.; Sun, C.; Busch, A.; Eken, S.M.; Saliba-Gustafsson, P.; Winter, H.; Winski, G.; Raaz, U.; et al. Local Delivery of miR-21 Stabilizes Fibrous Caps in Vulnerable Atherosclerotic Lesions. Mol. Ther. 2018, 26, 1040–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Zhu, L.; Huang, Y.; Zhang, Y.; Shen, W.; Fang, L.; Li, J.; Wang, Z.; Xie, Q. The relationship of MicroRNA-21 and plaque stability in acute coronary syndrome. Med. 2019, 98, e18049. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Narang, R.; Sreenivas, V.; Rastogi, V.; Bhatia, J.; Saluja, D.; Srivastava, K. Circulatory miR-133b and miR-21 as Novel Biomarkers in Early Prediction and Diagnosis of Coronary Artery Disease. Genes 2020, 11, 164. [Google Scholar] [CrossRef] [Green Version]
- Fazmin, I.T.; Achercouk, Z.; Edling, C.E.; Said, A.; Jeevaratnam, K. Circulating microRNA as a Biomarker for Coronary Artery Disease. Biomoleculs 2020, 10, 1354. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wang, E.; Wang, X.; Cong, X.; Chen, X. MicroRNA-21 is a unique signature associated with coronary plaque instability in humans by regulating matrix metalloproteinase-9 via reversion-inducing cysteine-rich protein with Kazal motifs. Exp. Mol. Pathol. 2014, 96, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Carino, A.; De Rosa, S.; Sorrentino, S.; Polimeni, A.; Sabatino, J.; Caiazzo, G.; Torella, D.; Spaccarotella, C.; Mongiardo, A.; Strangio, A.; et al. Modulation of Circulating MicroRNAs Levels during the Switch from Clopidogrel to Ticagrelor. BioMed Res. Int. 2016, 2016, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef] [PubMed]
- Izawa, H.; Amano, T. Plasma MicroRNA-100 as a Biomarker of Coronary Plaque Vulnerability. Circ. J. 2015, 79, 303–304. [Google Scholar] [CrossRef] [Green Version]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Kraczkowska, W.; Jagodziński, P.P. The Long Non-Coding RNA Landscape of Atherosclerotic Plaques. Mol. Diagn. Ther. 2019, 23, 735–749. [Google Scholar] [CrossRef] [Green Version]
- Mathy, N.W.; Chen, X.-M. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J. Biol. Chem. 2017, 292, 12375–12382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, J.; Scanlon, J.P.; Mahmoud, A.D.; Rodor, J.; Ballantyne, M.; Fontaine, M.A.; Temmerman, L.; Kaczynski, J.; Connor, K.L.; Bhushan, R.; et al. Novel Plaque Enriched Long Noncoding RNA in Atherosclerotic Macrophage Regulation (PELATON). Arter. Thromb. Vasc. Biol. 2020, 40, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Fan, Z.; Ma, J.; Liu, H.; Shen, L.; He, B.; Zhang, M. Profiling and functional characterization of circulation LncRNAs that are associated with coronary atherosclerotic plaque stability. Am. J. Transl Res 2019, 11, 3801–3815. [Google Scholar] [PubMed]
- Bourantas, C.V.; Garcia-Garcia, H.M.; Torii, R.; Zhang, Y.-J.; Westwood, M.; Crake, T.; Serruys, P.W. Vulnerable plaque detection: An unrealistic quest or a feasible objective with a clinical value? Hear. 2016, 102, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, K.; Kubo, T.; Akasaka, T. Evaluation of coronary plaques and atherosclerosis using optical coherence tomography. Expert Rev. Cardiovasc. Ther. 2021, 19, 379–386. [Google Scholar] [CrossRef]
- Su, M.-I.; Chen, C.-Y.; Yeh, H.-I.; Wang, K.-T. Concise Review of Optical Coherence Tomography in Clinical Practice. Acta Cardiol. Sin. 2016, 32, 381–386. [Google Scholar] [PubMed]
- Lv, R.; Maehara, A.; Matsumura, M.; Wang, L.; Wang, Q.; Zhang, C.; Guo, X.; Samady, H.; Giddens, D.P.; Zheng, J.; et al. Using optical coherence tomography and intravascular ultrasound imaging to quantify coronary plaque cap thickness and vulnerability: A pilot study. Biomed. Eng. Online 2020, 19, 1–18. [Google Scholar] [CrossRef]
- Bourantas, C.V.; Jaffer, F.A.; Gijsen, F.J.; van Soest, G.; Madden, S.P.; Courtney, B.K.; Fard, A.M.; Tenekecioglu, E.; Zeng, Y.; Van Der Steen, A.F.; et al. Hybrid intravascular imaging: Recent advances, technical considerations, and current applications in the study of plaque pathophysiology. Eur. Hear. J. 2017, 38, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Landmesser, U.; Galougahi, K.K.; Maehara, A.; Matsumura, M.; Shlofmitz, R.A.; Guagliumi, G.; Price, M.J.; Hill, J.M.; Akasaka, T.; et al. Optical coherence tomography-guided coronary stent implantation compared to angiography: A multicentre randomised trial in PCI – design and rationale of ILUMIEN IV: OPTIMAL PCI. EuroIntervention 2021, 16, 1092–1099. [Google Scholar] [CrossRef]
- Gardner, C.M.; Tan, H.; Hull, E.L.; Lisauskas, J.B.; Sum, S.T.; Meese, T.M.; Jiang, C.; Madden, S.P.; Caplan, J.D.; Burke, A.P.; et al. Detection of Lipid Core Coronary Plaques in Autopsy Specimens With a Novel Catheter-Based Near-Infrared Spectroscopy System. JACC: Cardiovasc. Imaging 2008, 1, 638–648. [Google Scholar] [CrossRef] [Green Version]
- Fard, A.M.; Vacas-Jacques, P.; Hamidi, E.; Wang, H.; Carruth, R.W.; Gardecki, J.A.; Tearney, G.J. Optical coherence tomography – near infrared spectroscopy system and catheter for intravascular imaging. Opt. Express 2013, 21, 30849–30858. [Google Scholar] [CrossRef] [Green Version]
- Steinvil, A.; Sadeh, B.; Arbel, Y.; Justo, D.; Belei, A.; Borenstein, N.; Banai, S.; Halkin, A. Prevalence and Predictors of Concomitant Carotid and Coronary Artery Atherosclerotic Disease. J. Am. Coll. Cardiol. 2011, 57, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Bots, M.L. Carotid intima-media thickness as a surrogate marker for cardiovascular disease in intervention studies. Curr. Med Res. Opin. 2006, 22, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, B.A.N.; Velema, E.; Schoneveld, A.H.; Vries, J.P.P.M.D.; De Bruin, P.; Seldenrijk, C.A.; Kleijn, D.P.V.D.; Busser, E.; Van Der Graaf, Y.; Moll, F.; et al. Athero-express: Differential atherosclerotic plaque expression of mRNA and protein in relation to cardiovascular events and patient characteristics. Rationale and design. Eur. J. Epidemiology 2004, 19, 1127–1133. [Google Scholar] [CrossRef]
- Hellings, W.E.; Moll, F.L.; De Kleijn, D.P.; Pasterkamp, G. 10-years experience with the Athero-Express study. Cardiovascular Diagnosis and Therapy 2012, 2, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Hellings, W.E.; Peeters, W.; Moll, F.L.; Piers, S.R.; van Setten, J.; Van der Spek, P.J.; de Vries, J.P.; Seldenrijk, K.A.; De Bruin, P.C.; Vink, A. Composition of carotid atherosclerotic plaque is associated with cardiovascular outcome: A prognostic study. Circulation 2010, 121, 1941–1950. [Google Scholar] [CrossRef] [Green Version]
- Ciccone, M.; Marzullo, A.; Mizio, D.; Angiletta, D.; Cortese, F.; Scicchitano, P.; Carbonara, S.; Ricci, G.; Regina, G.; Caruso, G.; et al. Can Carotid Plaque Histology Selectively Predict the Risk of an Acute Coronary Syndrome? Int. Hear. J. 2011, 52, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Staub, D.; Schinkel, A.F.; Coll, B.; Coli, S.; van der Steen, A.F.; Reed, J.D.; Krueger, C.; Thomenius, K.E.; Adam, D.; Sijbrands, E.J. Contrast-enhanced ultrasound imaging of the vasa vasorum: From early atherosclerosis to the identification of unstable plaques. JACC Cardiovasc. Imaging 2010, 3, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, B.; Hellings, W.E.; Moll, F.L.; de Vries, J.P.; de Kleijn, D.P.; de Bruin, P.; Busser, E.; Schoneveld, A.H.; Pasterkamp, G. Carotid atherosclerotic plaques in patients with transient ischemic attacks and stroke have unstable characteristics compared with plaques in asymptomatic and amaurosis fugax patients. J. Vasc. Surg. 2005, 42, 1075–1081. [Google Scholar] [CrossRef]
- Boekhorst, B.C.T.; Bovens, S.M.; Hellings, W.E.; Van Der Kraak, P.H.; Van De Kolk, K.W.; Vink, A.; Moll, F.L.; Van Oosterhout, M.F.; De Vries, J.P.; Doevendans, P.A.; et al. Molecular MRI of murine atherosclerotic plaque targeting NGAL: A protein associated with unstable human plaque characteristics. Cardiovasc. Res. 2010, 89, 680–688. [Google Scholar] [CrossRef] [Green Version]
- Derksen, W.J.; Peeters, W.; Van Lammeren, G.W.; Tersteeg, C.; De Vries, J.-P.P.; De Kleijn, D.P.; Moll, F.L.; Van Der Wal, A.C.; Pasterkamp, G.; Vink, A. Different stages of intraplaque hemorrhage are associated with different plaque phenotypes: A large histopathological study in 794 carotid and 276 femoral endarterectomy specimens. Atheroscler. 2011, 218, 369–377. [Google Scholar] [CrossRef]
- Ionita, M.G.; Borne, P.V.D.; Catanzariti, L.M.; Moll, F.L.; De Vries, J.-P.P.; Pasterkamp, G.; Vink, A.; De Kleijn, D.P.; De Vries, J.-P.P.M. High Neutrophil Numbers in Human Carotid Atherosclerotic Plaques Are Associated With Characteristics of Rupture-Prone Lesions. Arter. Thromb. Vasc. Biol. 2010, 30, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.; Eliasziw, M.; Gutnikov, S.; Warlow, C.; Barnett, H. Endarterectomy for symptomatic carotid stenosis in relation to clinical subgroups and timing of surgery. Lancet 2004, 363, 915–924. [Google Scholar] [CrossRef]
- Muntendam, P.; McCall, C.; Sanz, J.; Falk, E.; Fuster, V. The BioImage Study: Novel approaches to risk assessment in the primary prevention of atherosclerotic cardiovascular disease—study design and objectives. Am. Hear. J. 2010, 160, 49–57.e1. [Google Scholar] [CrossRef]
- Guo, X.; Maehara, A.; Matsumura, M.; Wang, L.; Zheng, J.; Samady, H.; Mintz, G.S.; Giddens, D.P.; Tang, D. Predicting plaque vulnerability change using intravascular ultrasound + optical coherence tomography image-based fluid–structure interaction models and machine learning methods with patient follow-up data: A feasibility study. Biomed. Eng. Online 2021, 20, 1–18. [Google Scholar] [CrossRef]
- Pan, J.; Cai, Y.; Wang, L.; Maehara, A.; Mintz, G.S.; Tang, D.; Li, Z. A prediction tool for plaque progression based on patient-specific multi-physical modeling. PLoS Comput. Biol. 2021, 17, e1008344. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Cai, Y.; He, C.; Li, Z. Coupled Modeling of Lipid Deposition, Inflammatory Response and Intraplaque Angiogenesis in Atherosclerotic Plaque. Ann. Biomed. Eng. 2018, 47, 439–452. [Google Scholar] [CrossRef]
- Guo, M.; Cai, Y.; Yao, X.; Li, Z. Mathematical modeling of atherosclerotic plaque destabilization: Role of neovascularization and intraplaque hemorrhage. J. Theor. Biol. 2018, 450, 53–65. [Google Scholar] [CrossRef] [PubMed]


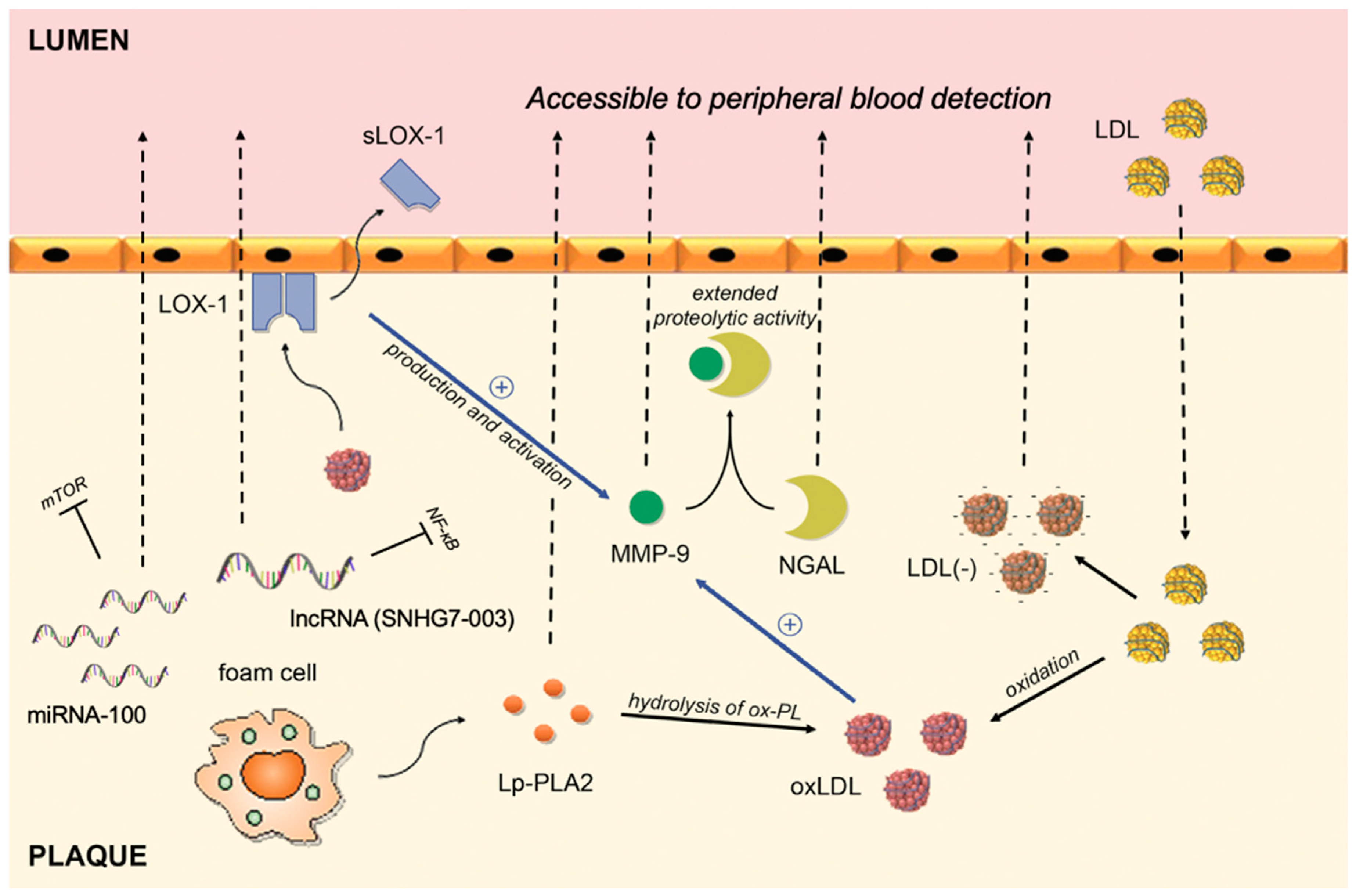{kind=link}
| Biomarker | Pathophysiological Pathway | Supporting Evidence |
|---|---|---|
| hs-CRP | An acute phase protein that, apart from liver, can be synthesized in plaques by macrophages or smooth muscle-like cells | Inoue et al. [65] Norja et al. [66] |
| MMP-9 | A proteolytic enzyme capable of degrading the extracellular matrix; upregulated in human macrophages stimulated by ox-LDL | Ezhov et al. [84] Wang et al. [85] |
| NGAL | Creates a complex with MMP-9 that inhibits degradation of MMP-9, thus extending its proteolytic activity | Eilenberg et al. [87] |
| sLOX-1 | A soluble form of LOX-1 receptor; interaction between ox-LDL (ligand) and LOX-1 (receptor) plays a role in vascular dysfunction | Hayashida et al. [100] Ueda et al. [101] Kobayashi et al. [102] |
| Ox-LDL | An oxidized fraction of the LDL; major participant in the proinflammatory processes associated with plaque rupture | Wang et al. [110] Wang et al. [111] Sigala et al. [112] |
| Electronegative LDL | A modified fraction of the LDL; physical and chemical characteristics differ from native LDL (increased Lp-PLA2 activity, ceramide, clusterine, non-esterified fatty acid content, increased aggregation level) | Lu et al. [123] Yang et al. [124] |
| Lp-PLA2 | An enzyme formed by macrophages and foam cells; hydrolyses oxidized phospholipids on LDL particles and subsequently releases proinflammatory lipids | Sarlon-Bartoli et al. [130] Dong-Ling et al. [131] Fenning et al. [135] |
| MicroRNA-100 | Post-transcriptional regulation | Soeki et al. [139] |
| MicroRNA-21 | Post-transcriptional regulation | Jin et al. [141] He et al. [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumric, M.; Borovac, J.A.; Martinovic, D.; Ticinovic Kurir, T.; Bozic, J. Circulating Biomarkers Reflecting Destabilization Mechanisms of Coronary Artery Plaques: Are We Looking for the Impossible? Biomolecules 2021, 11, 881. https://doi.org/10.3390/biom11060881
Kumric M, Borovac JA, Martinovic D, Ticinovic Kurir T, Bozic J. Circulating Biomarkers Reflecting Destabilization Mechanisms of Coronary Artery Plaques: Are We Looking for the Impossible? Biomolecules. 2021; 11(6):881. https://doi.org/10.3390/biom11060881
Chicago/Turabian StyleKumric, Marko, Josip A. Borovac, Dinko Martinovic, Tina Ticinovic Kurir, and Josko Bozic. 2021. "Circulating Biomarkers Reflecting Destabilization Mechanisms of Coronary Artery Plaques: Are We Looking for the Impossible?" Biomolecules 11, no. 6: 881. https://doi.org/10.3390/biom11060881
APA StyleKumric, M., Borovac, J. A., Martinovic, D., Ticinovic Kurir, T., & Bozic, J. (2021). Circulating Biomarkers Reflecting Destabilization Mechanisms of Coronary Artery Plaques: Are We Looking for the Impossible? Biomolecules, 11(6), 881. https://doi.org/10.3390/biom11060881