
Lipoprotein Lipase and Its Delivery of Fatty Acids to the Heart

{kind=link}
Abstract
:1. Introduction
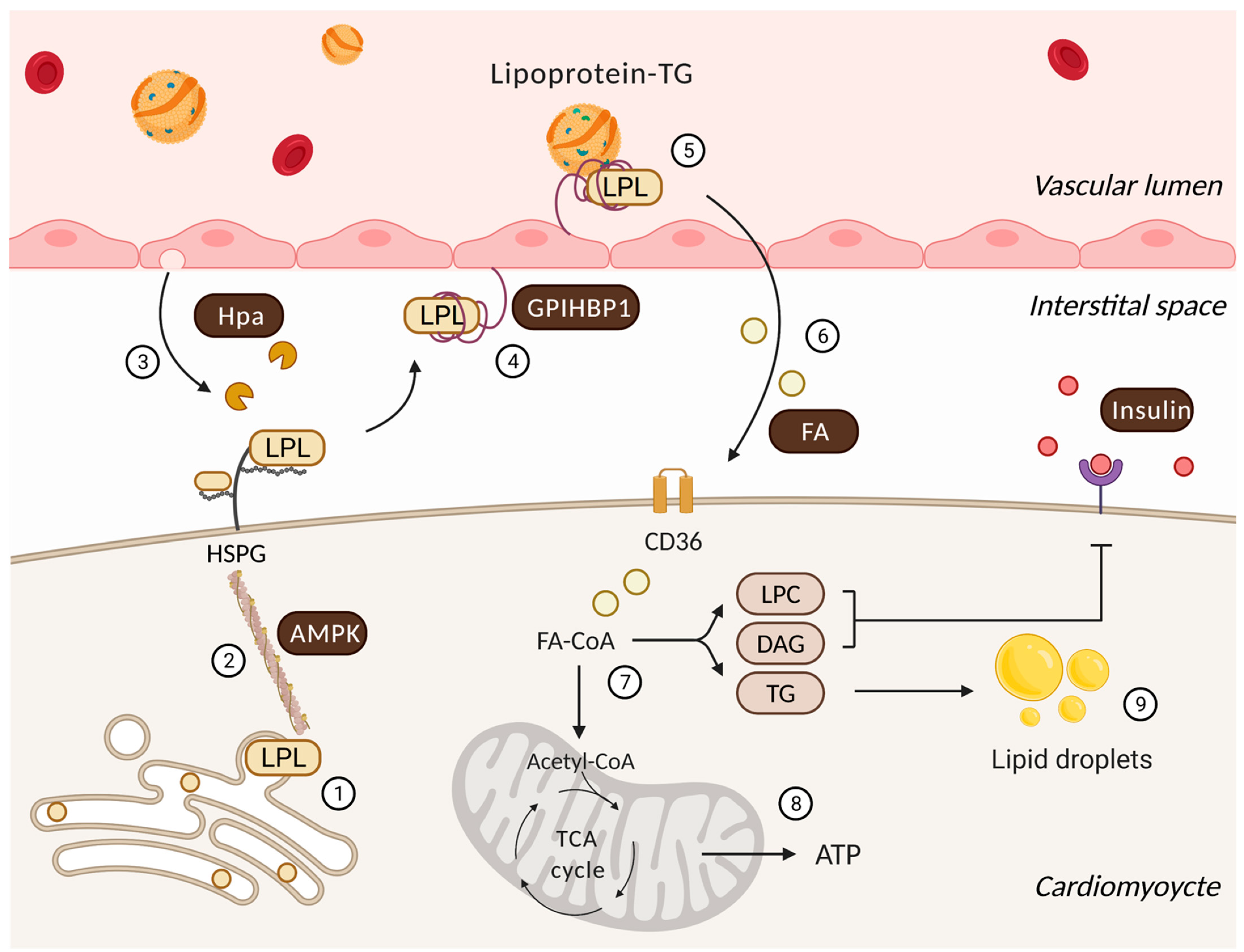
2. Cardiac Lipoprotein Lipase—Overview
3. Posttranslational Processes That Regulate Cardiac LPL
3.1. AMP-Activate Protein Kinase
3.2. Heparanase
3.3. GPIHBP1
3.4. Fatty Acid
3.5. Insulin
4. The Consequences of Oscillations in LPL
4.1. Gain-and Loss-of-Function of Cardiac LPL
4.2. Fluctuations in Cardiac LPL Following Diabetes
4.3. Lipid Metabolites and Diabetic Cardiomyopathy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Rodrigues, B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1489–H1506. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Wang, Y.; Rodrigues, B. Lipoprotein lipase mediated fatty acid delivery and its impact in diabetic cardiomyopathy. Biochim. Biophys. Acta 2012, 1821, 800–808. [Google Scholar] [CrossRef]
- Camps, L.; Reina, M.; Llobera, M.; Vilaro, S.; Olivecrona, T. Lipoprotein lipase: Cellular origin and functional distribution. Am. J. Physiol. 1990, 258, C673–C681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchette-Mackie, E.J.; Masuno, H.; Dwyer, N.K.; Olivecrona, T.; Scow, R.O. Lipoprotein lipase in myocytes and capillary endothelium of heart: Immunocytochemical study. Am. J. Physiol. 1989, 256, E818–E828. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.E.; Severson, D.L. Regulation of the synthesis, processing and translocation of lipoprotein lipase. Biochem. J. 1992, 287 Pt 2, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Auwerx, J.; Leroy, P.; Schoonjans, K. Lipoprotein lipase: Recent contributions from molecular biology. Crit. Rev. Clin. Lab. Sci. 1992, 29, 243–268. [Google Scholar] [CrossRef] [PubMed]
- Beigneux, A.P.; Allan, C.M.; Sandoval, N.P.; Cho, G.W.; Heizer, P.J.; Jung, R.S.; Stanhope, K.L.; Havel, P.J.; Birrane, G.; Meiyappan, M.; et al. Lipoprotein lipase is active as a monomer. Proc. Natl. Acad. Sci. USA 2019, 116, 6319–6328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, B.; Cam, M.C.; Jian, K.; Lim, F.; Sambandam, N.; Shepherd, G. Differential effects of streptozotocin-induced diabetes on cardiac lipoprotein lipase activity. Diabetes 1997, 46, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Enerback, S.; Gimble, J.M. Lipoprotein lipase gene expression: Physiological regulators at the transcriptional and post-transcriptional level. Biochim. Biophys. Acta 1993, 1169, 107–125. [Google Scholar] [CrossRef]
- Eckel, R.H. Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases. N. Engl. J. Med. 1989, 320, 1060–1068. [Google Scholar] [CrossRef]
- Allan, C.M.; Larsson, M.; Jung, R.S.; Ploug, M.; Bensadoun, A.; Beigneux, A.P.; Fong, L.G.; Young, S.G. Mobility of “HSPG-bound” LPL explains how LPL is able to reach GPIHBP1 on capillaries. J. Lipid Res. 2017, 58, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Ioka, R.X.; Kang, M.J.; Kamiyama, S.; Kim, D.H.; Magoori, K.; Kamataki, A.; Ito, Y.; Takei, Y.A.; Sasaki, M.; Suzuki, T.; et al. Expression cloning and characterization of a novel glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein, GPI-HBP1. J. Biol. Chem. 2003, 278, 7344–7349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.G.; Davies, B.S.; Voss, C.V.; Gin, P.; Weinstein, M.M.; Tontonoz, P.; Reue, K.; Bensadoun, A.; Fong, L.G.; Beigneux, A.P. GPIHBP1, an endothelial cell transporter for lipoprotein lipase. J. Lipid Res. 2011, 52, 1869–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.S.; Beigneux, A.P.; Barnes, R.H., 2nd; Tu, Y.; Gin, P.; Weinstein, M.M.; Nobumori, C.; Nyren, R.; Goldberg, I.; Olivecrona, G.; et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010, 12, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, M.M.; Yin, L.; Tu, Y.; Wang, X.; Wu, X.; Castellani, L.W.; Walzem, R.L.; Lusis, A.J.; Fong, L.G.; Beigneux, A.P.; et al. Chylomicronemia elicits atherosclerosis in mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 20–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.G.; Davies, B.S.; Fong, L.G.; Gin, P.; Weinstein, M.M.; Bensadoun, A.; Beigneux, A.P. GPIHBP1: An endothelial cell molecule important for the lipolytic processing of chylomicrons. Curr. Opin. Lipidol. 2007, 18, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, I.J.; Eckel, R.H.; McPherson, R. Triglycerides and heart disease: Still a hypothesis? Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1716–1725. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, K.G.; Hiyama, Y.; Hu, Y.; Huggins, L.A.; Ramakrishnan, R.; Abumrad, N.A.; Shulman, G.I.; Blaner, W.S.; Goldberg, I.J. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: In vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J. Biol. Chem. 2010, 285, 37976–37986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, N.; Barr, A.J.; Barr, R.L.; Desai, S.; Lopaschuk, G.D. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5’-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 1995, 270, 17513–17520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luiken, J.J.; Coort, S.L.; Willems, J.; Coumans, W.A.; Bonen, A.; van der Vusse, G.J.; Glatz, J.F. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 2003, 52, 1627–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, D.; Kewalramani, G.; Qi, D.; Pulinilkunnil, T.; Ghosh, S.; Abrahani, A.; Wambolt, R.; Allard, M.; Innis, S.M.; Rodrigues, B. beta-Agonist stimulation produces changes in cardiac AMPK and coronary lumen LPL only during increased workload. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1120–E1127. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Pulinilkunnil, T.; Qi, D.; Ghosh, S.; Abrahani, A.; Rodrigues, B. The metabolic “switch” AMPK regulates cardiac heparin-releasable lipoprotein lipase. Am. J. Physiology. Endocrinol. Metab. 2005, 288, E246–E253. [Google Scholar] [CrossRef] [Green Version]
- Puri, K.; Lal, N.; Shang, R.; Ghosh, S.; Flibotte, S.; Dyer, R.; Hussein, B.; Rodrigues, B. Diabetes mellitus severity and a switch from using lipoprotein lipase to adipose-derived fatty acid results in a cardiac metabolic signature that embraces cell death. J. Am. Heart Assoc. 2019, 8, e014022. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Song, P.; Xu, J.; Zhang, M.; Zou, M.H. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 9777–9788. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Puthanveetil, P.; Wang, F.; Kim, M.S.; Abrahani, A.; Rodrigues, B. Severity of diabetes governs vascular lipoprotein lipase by affecting enzyme dimerization and disassembly. Diabetes 2011, 60, 2041–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairbanks, M.B.; Mildner, A.M.; Leone, J.W.; Cavey, G.S.; Mathews, W.R.; Drong, R.F.; Slightom, J.L.; Bienkowski, M.J.; Smith, C.W.; Bannow, C.A.; et al. Processing of the human heparanase precursor and evidence that the active enzyme is a heterodimer. J. Biol. Chem. 1999, 274, 29587–29590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingis-Velitski, S.; Zetser, A.; Kaplan, V.; Ben-Zaken, O.; Cohen, E.; Levy-Adam, F.; Bashenko, Y.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J. Biol. Chem. 2004, 279, 44084–44092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikas, D.S.; Li, J.P.; Vlodavsky, I.; Lindahl, U. Substrate specificity of heparanases from human hepatoma and platelets. J. Biol. Chem. 1998, 273, 18770–18777. [Google Scholar] [CrossRef] [Green Version]
- Abboud-Jarrous, G.; Rangini-Guetta, Z.; Aingorn, H.; Atzmon, R.; Elgavish, S.; Peretz, T.; Vlodavsky, I. Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. J. Biol. Chem. 2005, 280, 13568–13575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zcharia, E.; Metzger, S.; Chajek-Shaul, T.; Aingorn, H.; Elkin, M.; Friedmann, Y.; Weinstein, T.; Li, J.P.; Lindahl, U.; Vlodavsky, I. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. FASEB J. 2004, 18, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Chiu, A.P.; Wan, A.; Neumaier, K.; Vlodavsky, I.; Rodrigues, B. Endothelial heparanase regulates heart metabolism by stimulating lipoprotein lipase secretion from cardiomyocytes. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 894–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafat, I.; Ilan, N.; Zoabi, S.; Vlodavsky, I.; Nakhoul, F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, A.; Van-Dijk, D.J.; Aingorn, H.; Erman, A.; Davies, M.; Darmon, D.; Hurvitz, H.; Vlodavsky, I. Involvement of human heparanase in the pathogenesis of diabetic nephropathy. Isr. Med Assoc. J. 2002, 4, 996–1002. [Google Scholar]
- Wang, F.; Kim, M.S.; Puthanveetil, P.; Kewalramani, G.; Deppe, S.; Ghosh, S.; Abrahani, A.; Rodrigues, B. Endothelial heparanase secretion after acute hypoinsulinemia is regulated by glucose and fatty acid. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1108–H1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, Y.; Kim, M.S.; Puthanveetil, P.; Ghosh, S.; Luciani, D.S.; Johnson, J.D.; Abrahani, A.; Rodrigues, B. Glucose-induced endothelial heparanase secretion requires cortical and stress actin reorganization. Cardiovasc. Res. 2010, 87, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fux, L.; Ilan, N.; Sanderson, R.D.; Vlodavsky, I. Heparanase: Busy at the cell surface. Trends Biochem. Sci. 2009, 34, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingis-Velitski, S.; Zetser, A.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J. Biol. Chem. 2004, 279, 23536–23541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.X.; Shao, C.H.; Liu, Q.; Yu, W.J.; Fang, J.P.; Yu, W.S.; Ali, A.; Ding, K. Heparanase enhances nerve-growth-factor-induced PC12 cell neuritogenesis via the p38 MAPK pathway. Biochem. J. 2011, 440, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, A.; Ilan, N.; Vlodavsky, I.; Li, J.P.; Johansson, S. Characterization of heparanase-induced phosphatidylinositol 3-kinase-AKT activation and its integrin dependence. J. Biol. Chem. 2013, 288, 12366–12375. [Google Scholar] [CrossRef] [Green Version]
- Chiu, A.P.; Wan, A.; Lal, N.; Zhang, D.; Wang, F.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Cardiomyocyte VEGF regulates endothelial cell GPIHBP1 to relocate lipoprotein lipase to the coronary lumen during diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigneux, A.P.; Davies, B.S.; Gin, P.; Weinstein, M.M.; Farber, E.; Qiao, X.; Peale, F.; Bunting, S.; Walzem, R.L.; Wong, J.S.; et al. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007, 5, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gin, P.; Yin, L.; Davies, B.S.; Weinstein, M.M.; Ryan, R.O.; Bensadoun, A.; Fong, L.G.; Young, S.G.; Beigneux, A.P. The acidic domain of GPIHBP1 is important for the binding of lipoprotein lipase and chylomicrons. J. Biol. Chem. 2008, 283, 29554–29562. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, K.K.; Midtgaard, S.R.; Mysling, S.; Kovrov, O.; Hansen, L.B.; Skar-Gislinge, N.; Beigneux, A.P.; Kragelund, B.B.; Olivecrona, G.; Young, S.G.; et al. A disordered acidic domain in GPIHBP1 harboring a sulfated tyrosine regulates lipoprotein lipase. Proc. Natl. Acad. Sci. USA 2018, 115, E6020–E6029. [Google Scholar] [CrossRef] [Green Version]
- Mysling, S.; Kristensen, K.K.; Larsson, M.; Kovrov, O.; Bensadouen, A.; Jorgensen, T.J.; Olivecrona, G.; Young, S.G.; Ploug, M. The angiopoietin-like protein ANGPTL4 catalyzes unfolding of the hydrolase domain in lipoprotein lipase and the endothelial membrane protein GPIHBP1 counteracts this unfolding. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.G.; Young, S.G.; Beigneux, A.P.; Bensadoun, A.; Oberer, M.; Jiang, H.; Ploug, M. GPIHBP1 and plasma triglyceride metabolism. Trends Endocrinol. Metab. 2016, 27, 455–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroupa, O.; Vorrsjo, E.; Stienstra, R.; Mattijssen, F.; Nilsson, S.K.; Sukonina, V.; Kersten, S.; Olivecrona, G.; Olivecrona, T. Linking nutritional regulation of Angptl4, Gpihbp1, and Lmf1 to lipoprotein lipase activity in rodent adipose tissue. BMC Physiol. 2012, 12, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei-Ling Chiu, A.; Wang, F.; Lal, N.; Wang, Y.; Zhang, D.; Hussein, B.; Wan, A.; Vlodavsky, I.; Rodrigues, B. Endothelial cells respond to hyperglycemia by increasing the LPL transporter GPIHBP1. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1274–E1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, A.P.; Bierende, D.; Lal, N.; Wang, F.; Wan, A.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Dual effects of hyperglycemia on endothelial cells and cardiomyocytes to enhance coronary LPL activity. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H82–H94. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Wang, F.; Puthanveetil, P.; Kewalramani, G.; Innis, S.; Marzban, L.; Steinberg, S.F.; Webber, T.D.; Kieffer, T.J.; Abrahani, A.; et al. Cleavage of protein kinase D after acute hypoinsulinemia prevents excessive lipoprotein lipase-mediated cardiac triglyceride accumulation. Diabetes 2009, 58, 2464–2475. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.; Bihain, B.E.; Bengtsson-Olivecrona, G.; Deckelbaum, R.J.; Carpentier, Y.A.; Olivecrona, T. Fatty acid control of lipoprotein lipase: A link between energy metabolism and lipid transport. Proc. Natl. Acad. Sci. USA 1990, 87, 909–913. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, G.; Olivecrona, T. Lipoprotein lipase. Mechanism of product inhibition. Eur. J. Biochem. 1980, 106, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Sukonina, V.; Lookene, A.; Olivecrona, T.; Olivecrona, G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. USA 2006, 103, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Burgess, S.C.; Ge, H.; Wong, K.K.; Nassem, R.H.; Garry, D.J.; Sherry, A.D.; Malloy, C.R.; Berger, J.P.; Li, C. Inhibition of cardiac lipoprotein utilization by transgenic overexpression of Angptl4 in the heart. Proc. Natl. Acad. Sci. USA 2005, 102, 1767–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafferty, M.J.; Bradford, K.C.; Erie, D.A.; Neher, S.B. Angiopoietin-like protein 4 inhibition of lipoprotein lipase: Evidence for reversible complex formation. J. Biol. Chem. 2013, 288, 28524–28534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koster, A.; Chao, Y.B.; Mosior, M.; Ford, A.; Gonzalez-DeWhitt, P.A.; Hale, J.E.; Li, D.; Qiu, Y.; Fraser, C.C.; Yang, D.D.; et al. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: Regulation of triglyceride metabolism. Endocrinology 2005, 146, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Yang, G.; Huang, L.; Motola, D.L.; Pourbahrami, T.; Li, C. Oligomerization and regulated proteolytic processing of angiopoietin-like protein 4. J. Biol. Chem. 2004, 279, 2038–2045. [Google Scholar] [CrossRef]
- Yin, W.; Romeo, S.; Chang, S.; Grishin, N.V.; Hobbs, H.H.; Cohen, J.C. Genetic variation in ANGPTL4 provides insights into protein processing and function. J. Biol. Chem. 2009, 284, 13213–13222. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, K.K.; Leth-Espensen, K.Z.; Mertens, H.D.T.; Birrane, G.; Meiyappan, M.; Olivecrona, G.; Jorgensen, T.J.D.; Young, S.G.; Ploug, M. Unfolding of monomeric lipoprotein lipase by ANGPTL4: Insight into the regulation of plasma triglyceride metabolism. Proc. Natl. Acad. Sci. USA 2020, 117, 4337–4346. [Google Scholar] [CrossRef] [PubMed]
- Mysling, S.; Kristensen, K.K.; Larsson, M.; Beigneux, A.P.; Gardsvoll, H.; Fong, L.G.; Bensadouen, A.; Jorgensen, T.J.; Young, S.G.; Ploug, M. The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. eLife 2016, 5, e12095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiadi, A.; Lichtenstein, L.; Degenhardt, T.; Boekschoten, M.V.; van Bilsen, M.; Desvergne, B.; Muller, M.; Kersten, S. Induction of cardiac Angptl4 by dietary fatty acids is mediated by peroxisome proliferator-activated receptor beta/delta and protects against fatty acid-induced oxidative stress. Circ. Res. 2010, 106, 1712–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staiger, H.; Haas, C.; Machann, J.; Werner, R.; Weisser, M.; Schick, F.; Machicao, F.; Stefan, N.; Fritsche, A.; Haring, H.U. Muscle-derived angiopoietin-like protein 4 is induced by fatty acids via peroxisome proliferator-activated receptor (PPAR)-delta and is of metabolic relevance in humans. Diabetes 2009, 58, 579–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Kewalramani, G.; Puthanveetil, P.; Lee, V.; Kumar, U.; An, D.; Abrahani, A.; Rodrigues, B. Acute diabetes moderates trafficking of cardiac lipoprotein lipase through p38 mitogen-activated protein kinase-dependent actin cytoskeleton organization. Diabetes 2008, 57, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Haller, J.F.; Mintah, I.J.; Shihanian, L.M.; Stevis, P.; Buckler, D.; Alexa-Braun, C.A.; Kleiner, S.; Banfi, S.; Cohen, J.C.; Hobbs, H.H.; et al. ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. J. Lipid Res. 2017, 58, 1166–1173. [Google Scholar] [CrossRef] [Green Version]
- Pulinilkunnil, T.; Abrahani, A.; Varghese, J.; Chan, N.; Tang, I.; Ghosh, S.; Kulpa, J.; Allard, M.; Brownsey, R.; Rodrigues, B. Evidence for rapid "metabolic switching" through lipoprotein lipase occupation of endothelial-binding sites. J. Mol. Cell. Cardiol. 2003, 35, 1093–1103. [Google Scholar] [CrossRef]
- Doolittle, M.H.; Ben-Zeev, O.; Elovson, J.; Martin, D.; Kirchgessner, T.G. The response of lipoprotein lipase to feeding and fasting. Evidence for posttranslational regulation. J. Biol. Chem. 1990, 265, 4570–4577. [Google Scholar] [CrossRef]
- Liu, G.; Olivecrona, T. Synthesis and transport of lipoprotein lipase in perfused guinea pig hearts. Am. J. Physiol. 1992, 263, H438–H446. [Google Scholar] [CrossRef]
- Hardie, D.G. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef]
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur. J. Biochem. 1997, 246, 259–273. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Schaffer, J.E. Lipotoxicity in the heart. Curr. Hypertens. Rep. 2005, 7, 412–417. [Google Scholar] [CrossRef] [PubMed]
- van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wende, A.R.; Symons, J.D.; Abel, E.D. Mechanisms of lipotoxicity in the cardiovascular system. Curr. Hypertens. Rep. 2012, 14, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levak-Frank, S.; Radner, H.; Walsh, A.; Stollberger, R.; Knipping, G.; Hoefler, G.; Sattler, W.; Weinstock, P.H.; Breslow, J.L.; Zechner, R. Muscle-specific overexpression of lipoprotein lipase causes a severe myopathy characterized by proliferation of mitochondria and peroxisomes in transgenic mice. J. Clin. Investig. 1995, 96, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Yagyu, H.; Chen, G.; Yokoyama, M.; Hirata, K.; Augustus, A.; Kako, Y.; Seo, T.; Hu, Y.; Lutz, E.P.; Merkel, M.; et al. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J. Clin. Investig. 2003, 111, 419–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulawa, L.K.; Eckel, R.H. Overexpression of muscle lipoprotein lipase and insulin sensitivity. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O.; Guo, X.; Taylor, K.D.; Quinones, M.J.; Saad, M.F.; Yang, H.; Hsueh, W.A.; Rotter, J.I. Lipoprotein lipase is a gene for insulin resistance in Mexican Americans. Diabetes 2004, 53, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Noh, H.L.; Okajima, K.; Molkentin, J.D.; Homma, S.; Goldberg, I.J. Acute lipoprotein lipase deletion in adult mice leads to dyslipidemia and cardiac dysfunction. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E755–E760. [Google Scholar] [CrossRef]
- Augustus, A.S.; Buchanan, J.; Park, T.S.; Hirata, K.; Noh, H.L.; Sun, J.; Homma, S.; D’Armiento, J.; Abel, E.D.; Goldberg, I.J. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J. Biol. Chem. 2006, 281, 8716–8723. [Google Scholar] [CrossRef] [Green Version]
- Kashiwazaki, K.; Hirano, T.; Yoshino, G.; Kurokawa, M.; Tajima, H.; Adachi, M. Decreased release of lipoprotein lipase is associated with vascular endothelial damage in NIDDM patients with microalbuminuria. Diabetes Care 1998, 21, 2016–2020. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Nikkila, E.A. Lipoprotein lipase activity of adipose tissue and skeletal muscle in insulin-deficient human diabetes. Relation to high-density and very-low-density lipoproteins and response to treatment. Diabetologia 1979, 17, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; Pulinilkunnil, T.; An, D.; Ghosh, S.; Abrahani, A.; Pospisilik, J.A.; Brownsey, R.; Wambolt, R.; Allard, M.; Rodrigues, B. Single-dose dexamethasone induces whole-body insulin resistance and alters both cardiac fatty acid and carbohydrate metabolism. Diabetes 2004, 53, 1790–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kewalramani, G.; Puthanveetil, P.; Kim, M.S.; Wang, F.; Lee, V.; Hau, N.; Beheshti, E.; Ng, N.; Abrahani, A.; Rodrigues, B. Acute dexamethasone-induced increase in cardiac lipoprotein lipase requires activation of both Akt and stress kinases. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E137–E147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambandam, N.; Abrahani, M.A.; St Pierre, E.; Al-Atar, O.; Cam, M.C.; Rodrigues, B. Localization of lipoprotein lipase in the diabetic heart: Regulation by acute changes in insulin. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1526–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambandam, N.; Abrahani, M.A.; Craig, S.; Al-Atar, O.; Jeon, E.; Rodrigues, B. Metabolism of VLDL is increased in streptozotocin-induced diabetic rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1874–H1882. [Google Scholar] [CrossRef]
- Pulinilkunnil, T.; Qi, D.; Ghosh, S.; Cheung, C.; Yip, P.; Varghese, J.; Abrahani, A.; Brownsey, R.; Rodrigues, B. Circulating triglyceride lipolysis facilitates lipoprotein lipase translocation from cardiomyocyte to myocardial endothelial lining. Cardiovasc. Res. 2003, 59, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Pulinilkunnil, T.; An, D.; Yip, P.; Chan, N.; Qi, D.; Ghosh, S.; Abrahani, A.; Rodrigues, B. Palmitoyl lysophosphatidylcholine mediated mobilization of LPL to the coronary luminal surface requires PKC activation. J. Mol. Cell. Cardiol. 2004, 37, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Wan, A.; Rodrigues, B. Endothelial cell-cardiomyocyte crosstalk in diabetic cardiomyopathy. Cardiovasc. Res. 2016, 111, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kewalramani, G.; An, D.; Kim, M.S.; Ghosh, S.; Qi, D.; Abrahani, A.; Pulinilkunnil, T.; Sharma, V.; Wambolt, R.B.; Allard, M.F.; et al. AMPK control of myocardial fatty acid metabolism fluctuates with the intensity of insulin-deficient diabetes. J. Mol. Cell. Cardiol. 2007, 42, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Wang, F.; Puthanveetil, P.; Kewalramani, G.; Hosseini-Beheshti, E.; Ng, N.; Wang, Y.; Kumar, U.; Innis, S.; Proud, C.G.; et al. Protein kinase D is a key regulator of cardiomyocyte lipoprotein lipase secretion after diabetes. Circ. Res. 2008, 103, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Pulinilkunnil, T.; An, D.; Ghosh, S.; Qi, D.; Kewalramani, G.; Yuen, G.; Virk, N.; Abrahani, A.; Rodrigues, B. Lysophosphatidic acid-mediated augmentation of cardiomyocyte lipoprotein lipase involves actin cytoskeleton reorganization. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2802–H2810. [Google Scholar] [CrossRef]
- Wang, Y.; Chiu, A.P.; Neumaier, K.; Wang, F.; Zhang, D.; Hussein, B.; Lal, N.; Wan, A.; Liu, G.; Vlodavsky, I.; et al. Endothelial cell heparanase taken up by cardiomyocytes regulates lipoprotein lipase transfer to the coronary lumen after diabetes. Diabetes 2014, 63, 2643–2655. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wan, A.; Chiu, A.P.; Wang, Y.; Wang, F.; Neumaier, K.; Lal, N.; Bround, M.J.; Johnson, J.D.; Vlodavsky, I.; et al. Hyperglycemia-induced secretion of endothelial heparanase stimulates a vascular endothelial growth factor autocrine network in cardiomyocytes that promotes recruitment of lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2830–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, N.; Chiu, A.P.; Wang, F.; Zhang, D.; Jia, J.; Wan, A.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Loss of VEGFB and its signaling in the diabetic heart is associated with increased cell death signaling. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1163–H1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, D.; Kuo, K.H.; Abrahani, A.; An, D.; Qi, Y.; Heung, J.; Kewalramani, G.; Pulinilkunnil, T.; Ghosh, S.; Innis, S.M.; et al. Acute intralipid infusion reduces cardiac luminal lipoprotein lipase but recruits additional enzyme from cardiomyocytes. Cardiovasc. Res. 2006, 72, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Regan, T.J.; Ahmed, S.; Haider, B.; Moschos, C.; Weisse, A. Diabetic cardiomyopathy: Experimental and clinical observations. N. J. Med. 1994, 91, 776–778. [Google Scholar] [PubMed]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 2010, 11, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.Y.; Prins, J.B.; Marwick, T.H. Diabetic cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Endocr. Rev. 2004, 25, 543–567. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Rodent models of diabetic cardiomyopathy. Dis. Model. Mech. 2009, 2, 454–466. [Google Scholar] [CrossRef] [Green Version]
- Severson, D.L. Diabetic cardiomyopathy: Recent evidence from mouse models of type 1 and type 2 diabetes. Can. J. Physiol. Pharmacol. 2004, 82, 813–823. [Google Scholar] [CrossRef]
- Shehadeh, A.; Regan, T.J. Cardiac consequences of diabetes mellitus. Clin. Cardiol. 1995, 18, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Fein, F.S.; Sonnenblick, E.H. Diabetic cardiomyopathy. Prog. Cardiovasc. Dis. 1985, 27, 255–270. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Liu, X.; Panagia, V.; Takeda, N. Subcellular remodeling and heart dysfunction in chronic diabetes. Cardiovasc. Res. 1998, 40, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Erion, D.M.; Shulman, G.I. Diacylglycerol-mediated insulin resistance. Nat. Med. 2010, 16, 400–402. [Google Scholar] [CrossRef] [Green Version]
- Han, M.S.; Lim, Y.M.; Quan, W.; Kim, J.R.; Chung, K.W.; Kang, M.; Kim, S.; Park, S.Y.; Han, J.S.; Park, S.Y.; et al. Lysophosphatidylcholine as an effector of fatty acid-induced insulin resistance. J. Lipid Res. 2011, 52, 1234–1246. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, R.; Rodrigues, B. Lipoprotein Lipase and Its Delivery of Fatty Acids to the Heart. Biomolecules 2021, 11, 1016. https://doi.org/10.3390/biom11071016
Shang R, Rodrigues B. Lipoprotein Lipase and Its Delivery of Fatty Acids to the Heart. Biomolecules. 2021; 11(7):1016. https://doi.org/10.3390/biom11071016
Chicago/Turabian StyleShang, Rui, and Brian Rodrigues. 2021. "Lipoprotein Lipase and Its Delivery of Fatty Acids to the Heart" Biomolecules 11, no. 7: 1016. https://doi.org/10.3390/biom11071016
APA StyleShang, R., & Rodrigues, B. (2021). Lipoprotein Lipase and Its Delivery of Fatty Acids to the Heart. Biomolecules, 11(7), 1016. https://doi.org/10.3390/biom11071016