
Osteopontin in Cardiovascular Diseases

{kind=link}
{kind=link}
Abstract
:1. Introduction
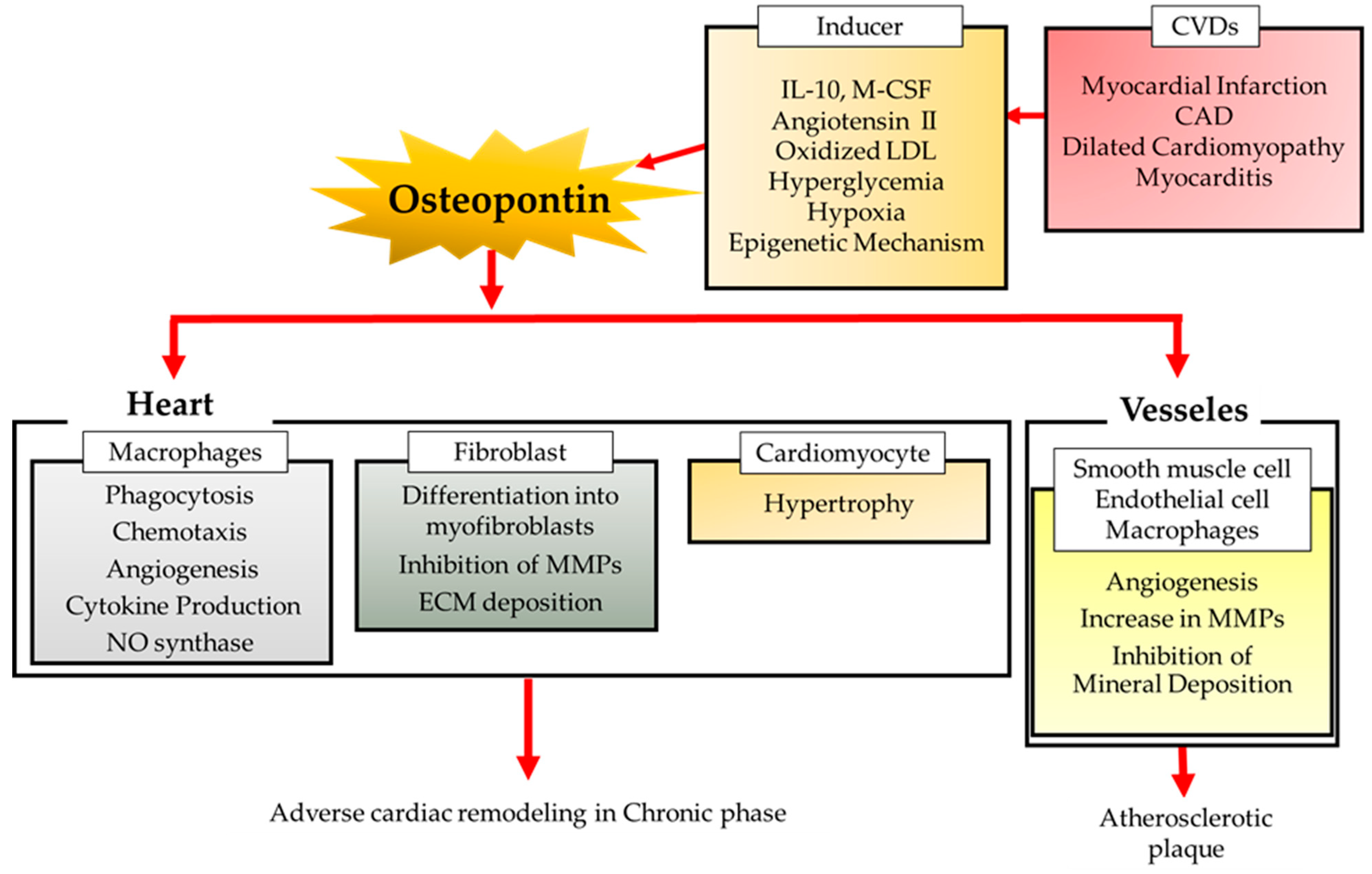
2. Diversity of the Origin and Regulation of OPN in CVDs
2.1. The Source of OPN
2.2. Inducer of OPN
2.3. OPN Isoforms
3. OPN in CVDs
3.1. Ischemic Heart Diseases
3.2. Hypertension
3.3. Heart Failure
3.4. Dilated Cardiomyopathy
3.5. Atherosclerosis
3.6. Cardiovascular Calcification
3.7. Other Heart Diseases
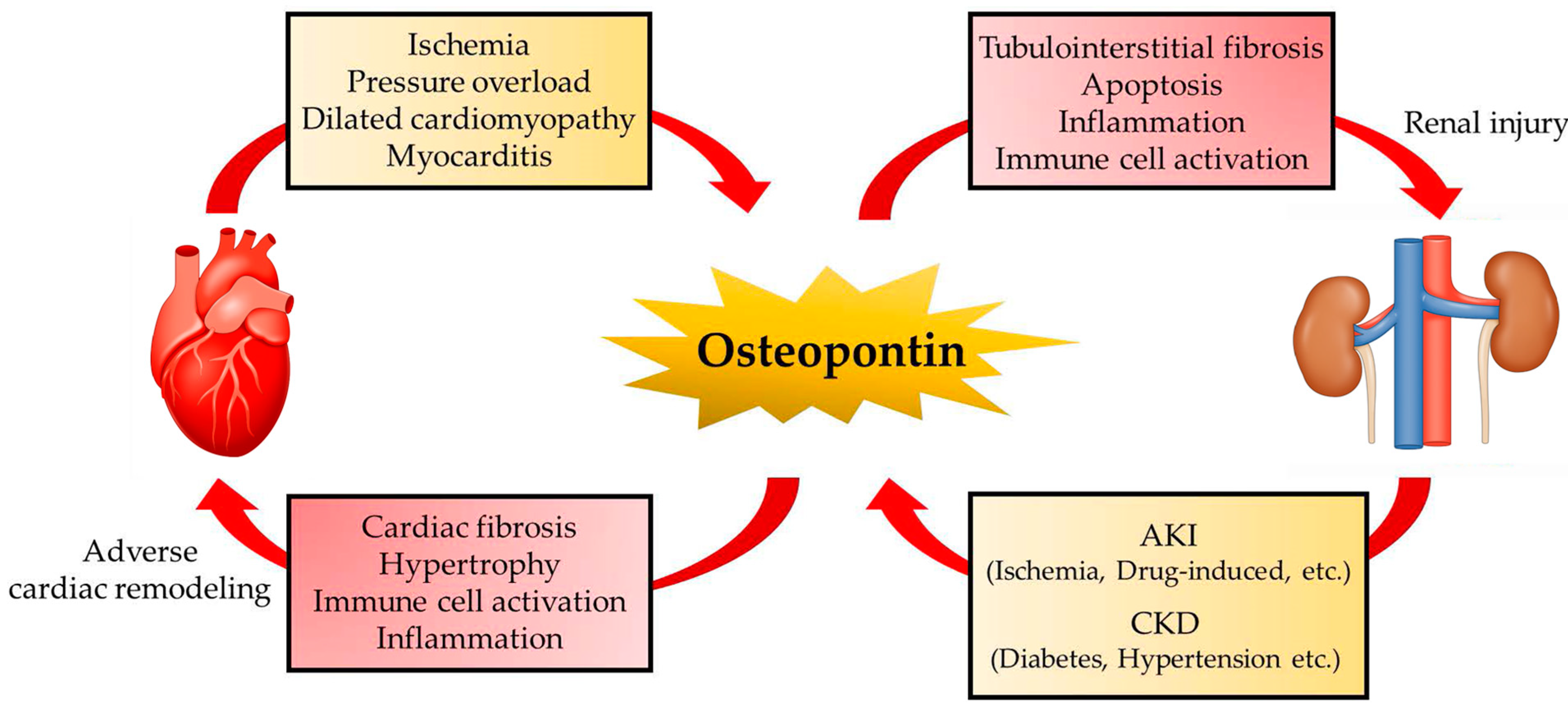
4. OPN in Multi-Organ Crosstalk
4.1. OPN in the Kidney
4.1.1. Acute Kidney Injury
4.1.2. Chronic Kidney Injury
4.2. Diabetes
4.3. Cardiorenal Syndrome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhindsa, D.S.; Sandesara, P.B.; Shapiro, M.D.; Wong, N.D. The Evolving Understanding and Approach to Residual Cardiovascular Risk Management. Front. Cardiovasc. Med. 2020, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e563–e595. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.K. Residual inflammatory risk: Lessons from trials for the future. Atherosclerosis 2020, 311, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Hervás, S.; González-Navarro, H. Anti-inflammatory Therapies for Cardiovascular Disease: Signaling Pathways and Mechanisms. Rev. Esp. Cardiol. 2019, 72, 767–773. [Google Scholar] [CrossRef]
- Lutgens, E.; Atzler, D.; Döring, Y.; Duchene, J.; Steffens, S.; Weber, C. Immunotherapy for cardiovascular disease. Eur. Heart J. 2019, 40, 3937–3946. [Google Scholar] [CrossRef]
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and cardiovascular disease: From pathogenesis to therapeutic target. Curr. Atheroscler. Rep. 2014, 16, 435. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, O.; Mohanty, B.D.; Martin, S.S.; Joshi, P.H.; Blaha, M.J.; Nasir, K.; Blumenthal, R.S.; Budoff, M.J. High-sensitivity C-reactive protein and cardiovascular disease: A resolute belief or an elusive link? J. Am. Coll. Cardiol. 2013, 62, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Sattar, N.; Murray, H.M.; McConnachie, A.; Blauw, G.J.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; et al. C-reactive protein and prediction of coronary heart disease and global vascular events in the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER). Circulation 2007, 115, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 2003, 107, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opstal, T.S.J.; Hoogeveen, R.M.; Fiolet, A.T.L.; Silvis, M.J.M.; The, S.H.K.; Bax, W.A.; de Kleijn, D.P.V.; Mosterd, A.; Stroes, E.S.G.; Cornel, J.H. Colchicine Attenuates Inflammation Beyond the Inflammasome in Chronic Coronary Artery Disease: A LoDoCo2 Proteomic Substudy. Circulation 2020, 142, 1996–1998. [Google Scholar] [CrossRef] [PubMed]
- Lok, Z.S.Y.; Lyle, A.N. Osteopontin in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelaziz Mohamed, I.; Gadeau, A.P.; Hasan, A.; Abdulrahman, N.; Mraiche, F. Osteopontin: A Promising Therapeutic Target in Cardiac Fibrosis. Cells 2019, 8, 1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waller, A.H.; Sanchez-Ross, M.; Kaluski, E.; Klapholz, M. Osteopontin in cardiovascular disease: A potential therapeutic target. Cardiol. Rev. 2010, 18, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Lamort, A.S.; Giopanou, I.; Psallidas, I.; Stathopoulos, G.T. Osteopontin as a Link between Inflammation and Cancer: The Thorax in the Spotlight. Cells 2019, 8, 815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaleta, B. The role of osteopontin in kidney diseases. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2019, 68, 93–102. [Google Scholar] [CrossRef]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Cho, H.J.; Kim, H.S. Osteopontin: A multifunctional protein at the crossroads of inflammation, atherosclerosis, and vascular calcification. Curr. Atheroscler. Rep. 2009, 11, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Wang, C.; Zhang, D.; Wang, B.; Zhou, Y. Osteopontin in Bone Metabolism and Bone Diseases. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e919159. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M.; Steitz, S. Osteopontin: A versatile regulator of inflammation and biomineralization. Matrix Biol. J. Int. Soc. Matrix Biol. 2000, 19, 615–622. [Google Scholar] [CrossRef]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yoshida, N.; Yamamoto, T.; Isobe, S.; Moriyama, H.; Goto, S.; Kitakata, H.; et al. IL (Interleukin)-10-STAT3-Galectin-3 Axis Is Essential for Osteopontin-Producing Reparative Macrophage Polarization After Myocardial Infarction. Circulation 2018, 138, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Li, N.Y.; Wai, P.Y.; Kuo, P.C. Epithelial-mesenchymal transition, TGF-β, and osteopontin in wound healing and tissue remodeling after injury. J. Burn Care Res. Off. Publ. Am. Burn Assoc. 2012, 33, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, M.D.; Pedraza, C.E.; Kaartinen, M.T. Osteopontin and wound healing in bone. Cells Tissues Organs 2011, 194, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Foster, C.R.; Dalal, S.; Singh, K. Osteopontin: Role in extracellular matrix deposition and myocardial remodeling post-MI. J. Mol. Cell. Cardiol. 2010, 48, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, K.; Sano, M. Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions. Int. J. Mol. Sci. 2020, 21, 7676. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Yan, X.; Shinmura, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yamamoto, T.; Anzai, A.; Isobe, S.; Yoshida, N.; et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J. Clin. Investig. 2016, 126, 4626–4639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalrhim, A.D.; Marroush, T.S.; Austin, E.E.; Gersh, B.J.; Solak, N.; Rizvi, S.A.; Bailey, K.R.; Kullo, I.J. Plasma Osteopontin Levels and Adverse Cardiovascular Outcomes in the PEACE Trial. PLoS ONE 2016, 11, e0156965. [Google Scholar] [CrossRef] [PubMed]
- Klingel, K.; Kandolf, R. Osteopontin: A biomarker to predict the outcome of inflammatory heart disease. Semin. Thromb. Hemost. 2010, 36, 195–202. [Google Scholar] [CrossRef]
- Yousefi, K.; Irion, C.I.; Takeuchi, L.M.; Ding, W.; Lambert, G.; Eisenberg, T.; Sukkar, S.; Granzier, H.L.; Methawasin, M.; Lee, D.I.; et al. Osteopontin Promotes Left Ventricular Diastolic Dysfunction Through a Mitochondrial Pathway. J. Am. Coll. Cardiol. 2019, 73, 2705–2718. [Google Scholar] [CrossRef] [PubMed]
- Trueblood, N.A.; Xie, Z.; Communal, C.; Sam, F.; Ngoy, S.; Liaw, L.; Jenkins, A.W.; Wang, J.; Sawyer, D.B.; Bing, O.H.; et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ. Res. 2001, 88, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalay, G.; Sauter, M.; Haberland, M.; Zuegel, U.; Steinmeyer, A.; Kandolf, R.; Klingel, K. Osteopontin: A fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ. Res. 2009, 104, 851–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, F.; Xie, Z.; Ooi, H.; Kerstetter, D.L.; Colucci, W.S.; Singh, M.; Singh, K. Mice lacking osteopontin exhibit increased left ventricular dilation and reduced fibrosis after aldosterone infusion. Am. J. Hypertens. 2004, 17, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Sirokman, G.; Communal, C.; Robinson, K.G.; Conrad, C.H.; Brooks, W.W.; Bing, O.H.; Colucci, W.S. Myocardial osteopontin expression coincides with the development of heart failure. Hypertension 1999, 33, 663–670. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.B.; Halpert, I.; Wickline, S.; Davison, G.; Parks, W.C.; Rottman, J.N. Osteopontin expression is increased in the heritable cardiomyopathy of Syrian hamsters. Circulation 1995, 92, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Rigamonti, F.; Burger, F.; Roth, A.; Bertolotto, M.; Spinella, G.; Pane, B.; Palombo, D.; Pende, A.; Bonaventura, A.; et al. Serum levels of osteopontin predict major adverse cardiovascular events in patients with severe carotid artery stenosis. Int. J. Cardiol. 2018, 255, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, M.; Pedersen, S.H.; Møgelvang, R.; Lindberg, S.; Jensen, J.S.; Galatius, S.; Flyvbjerg, A. High osteopontin levels predict long-term outcome after STEMI and primary percutaneous coronary intervention. Eur. J. Prev. Cardiol. 2013, 20, 922–929. [Google Scholar] [CrossRef]
- Murry, C.E.; Giachelli, C.M.; Schwartz, S.M.; Vracko, R. Macrophages express osteopontin during repair of myocardial necrosis. Am. J. Pathol. 1994, 145, 1450–1462. [Google Scholar] [PubMed]
- Rittling, S.R. Osteopontin in macrophage function. Expert Rev. Mol. Med. 2011, 13, e15. [Google Scholar] [CrossRef]
- Schack, L.; Stapulionis, R.; Christensen, B.; Kofod-Olsen, E.; Skov Sørensen, U.B.; Vorup-Jensen, T.; Sørensen, E.S.; Höllsberg, P. Osteopontin enhances phagocytosis through a novel osteopontin receptor, the alphaXbeta2 integrin. J. Immunol. 2009, 182, 6943–6950. [Google Scholar] [CrossRef] [Green Version]
- Weber, G.F.; Zawaideh, S.; Hikita, S.; Kumar, V.A.; Cantor, H.; Ashkar, S. Phosphorylation-dependent interaction of osteopontin with its receptors regulates macrophage migration and activation. J. Leukoc. Biol. 2002, 72, 752–761. [Google Scholar] [PubMed]
- Lund, S.A.; Wilson, C.L.; Raines, E.W.; Tang, J.; Giachelli, C.M.; Scatena, M. Osteopontin mediates macrophage chemotaxis via α4 and α9 integrins and survival via the α4 integrin. J. Cell. Biochem. 2013, 114, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.; Peterson, J.T.; Subramanian, V.; Singh, M.; Singh, K. Inhibition of matrix metalloproteinases improves left ventricular function in mice lacking osteopontin after myocardial infarction. Mol. Cell. Biochem. 2009, 322, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Zheng, Y.H.; Tian, C.; Meng, Y.; Qin, Y.W.; Du, Y.H.; Du, J.; Li, H.H. Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells. J. Cell. Physiol. 2012, 227, 127–135. [Google Scholar] [CrossRef]
- Toyonaga, T.; Nakase, H.; Ueno, S.; Matsuura, M.; Yoshino, T.; Honzawa, Y.; Itou, A.; Namba, K.; Minami, N.; Yamada, S.; et al. Osteopontin Deficiency Accelerates Spontaneous Colitis in Mice with Disrupted Gut Microbiota and Macrophage Phagocytic Activity. PLoS ONE 2015, 10, e0135552. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kim, H.L.; Choi, J.S.; Choi, J.Y.; Cha, J.H.; Lee, M.Y. Osteopontin: Correlation with phagocytosis by brain macrophages in a rat model of stroke. Glia 2011, 59, 413–423. [Google Scholar] [CrossRef]
- Lenga, Y.; Koh, A.; Perera, A.S.; McCulloch, C.A.; Sodek, J.; Zohar, R. Osteopontin expression is required for myofibroblast differentiation. Circ. Res. 2008, 102, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Graf, K.; Do, Y.S.; Ashizawa, N.; Meehan, W.P.; Giachelli, C.M.; Marboe, C.C.; Fleck, E.; Hsueh, W.A. Myocardial osteopontin expression is associated with left ventricular hypertrophy. Circulation 1997, 96, 3063–3071. [Google Scholar] [CrossRef]
- Xie, Z.; Singh, M.; Singh, K. Osteopontin modulates myocardial hypertrophy in response to chronic pressure overload in mice. Hypertension 2004, 44, 826–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, V.; Krishnamurthy, P.; Singh, K.; Singh, M. Lack of osteopontin improves cardiac function in streptozotocin-induced diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H673–H683. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [Green Version]
- Burke, R.M.; Burgos Villar, K.N.; Small, E.M. Fibroblast contributions to ischemic cardiac remodeling. Cell. Signal. 2021, 77, 109824. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zohar, R.; Zhu, B.; Liu, P.; Sodek, J.; McCulloch, C.A. Increased cell death in osteopontin-deficient cardiac fibroblasts occurs by a caspase-3-independent pathway. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1730–H1739. [Google Scholar] [CrossRef]
- Ashizawa, N.; Graf, K.; Do, Y.S.; Nunohiro, T.; Giachelli, C.M.; Meehan, W.P.; Tuan, T.L.; Hsueh, W.A. Osteopontin is produced by rat cardiac fibroblasts and mediates A(II)-induced DNA synthesis and collagen gel contraction. J. Clin. Investig. 1996, 98, 2218–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, E.R.; Garvin, M.R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M.; Giachelli, C.M. Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 1648–1656. [Google Scholar] [CrossRef] [Green Version]
- Kusuyama, T.; Yoshiyama, M.; Omura, T.; Nishiya, D.; Enomoto, S.; Matsumoto, R.; Izumi, Y.; Akioka, K.; Takeuchi, K.; Iwao, H.; et al. Angiotensin blockade inhibits osteopontin expression in non-infarcted myocardium after myocardial infarction. J. Pharmacol. Sci. 2005, 98, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Pimental, D.R.; Lohan, S.; Vasertriger, A.; Pligavko, C.; Colucci, W.S.; Singh, K. Regulation of angiotensin II-stimulated osteopontin expression in cardiac microvascular endothelial cells: Role of p42/44 mitogen-activated protein kinase and reactive oxygen species. J. Cell. Physiol. 2001, 188, 132–138. [Google Scholar] [CrossRef]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Anzai, A.; Moriyama, H.; Kitakata, H.; Hiraide, T.; Ko, S.; Goto, S.; et al. MerTK Expression and ERK Activation Are Essential for the Functional Maturation of Osteopontin-Producing Reparative Macrophages After Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e017071. [Google Scholar] [CrossRef]
- Lorenzen, J.M.; Neunhöffer, H.; David, S.; Kielstein, J.T.; Haller, H.; Fliser, D. Angiotensin II receptor blocker and statins lower elevated levels of osteopontin in essential hypertension—Results from the EUTOPIA trial. Atherosclerosis 2010, 209, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Phadke, S.A.; Batlle, D.; Sahai, A. Hypoxia stimulates osteopontin expression and proliferation of cultured vascular smooth muscle cells: Potentiation by high glucose. Diabetes 2001, 50, 1482–1490. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ren, Y.; Kang, L.; Zhang, L. Oxidized low-density lipoprotein increases the proliferation and migration of human coronary artery smooth muscle cells through the upregulation of osteopontin. Int. J. Mol. Med. 2014, 33, 1341–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Balligand, J.L.; Fischer, T.A.; Smith, T.W.; Kelly, R.A. Glucocorticoids increase osteopontin expression in cardiac myocytes and microvascular endothelial cells. Role in regulation of inducible nitric oxide synthase. J. Biol. Chem. 1995, 270, 28471–28478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimba, E.R.; Tilli, T.M. Human osteopontin splicing isoforms: Known roles, potential clinical applications and activated signaling pathways. Cancer Lett. 2013, 331, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Suezawa, C.; Kusachi, S.; Murakami, T.; Toeda, K.; Hirohata, S.; Nakamura, K.; Yamamoto, K.; Koten, K.; Miyoshi, T.; Shiratori, Y. Time-dependent changes in plasma osteopontin levels in patients with anterior-wall acute myocardial infarction after successful reperfusion: Correlation with left-ventricular volume and function. J. Lab. Clin. Med. 2005, 145, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Shingai, M.; Aso, N.; Hazuku, T.; Nasu, M. Osteopontin is released from the heart into the coronary circulation in patients with a previous anterior wall myocardial infarction. Circ. J. Off. J. Jpn. Circ. Soc. 2003, 67, 742–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniatis, K.; Siasos, G.; Oikonomou, E.; Vavuranakis, M.; Zaromytidou, M.; Mourouzis, K.; Paraskevopoulos, T.; Charalambous, G.; Papavassiliou, A.G.; Tousoulis, D. Osteoprotegerin and Osteopontin Serum Levels are Associated with Vascular Function and Inflammation in Coronary Artery Disease Patients. Curr. Vasc. Pharmacol. 2020, 18, 523–530. [Google Scholar] [CrossRef]
- Georgiadou, P.; Iliodromitis, E.K.; Kolokathis, F.; Varounis, C.; Gizas, V.; Mavroidis, M.; Capetanaki, Y.; Boudoulas, H.; Kremastinos, D.T. Osteopontin as a novel prognostic marker in stable ischaemic heart disease: A 3-year follow-up study. Eur. J. Clin. Investig. 2010, 40, 288–293. [Google Scholar] [CrossRef]
- Spinale, F.G. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Mann, D.L.; Entman, M.L.; Spinale, F.G. Extracellular matrix remodeling following myocardial injury. Ann. Med. 2003, 35, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Mujumdar, V.S.; Smiley, L.M.; Tyagi, S.C. Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength. Int. J. Cardiol. 2001, 79, 277–286. [Google Scholar] [CrossRef]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Singh, M.; Siwik, D.A.; Joyner, W.L.; Singh, K. Osteopontin inhibits interleukin-1beta-stimulated increases in matrix metalloproteinase activity in adult rat cardiac fibroblasts: Role of protein kinase C-zeta. J. Biol. Chem. 2003, 278, 48546–48552. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.Y.; Bergman, M.R.; Nguyen, A.P.; Turcato, S.; Swigart, P.M.; Rodrigo, M.C.; Simpson, P.C.; Karliner, J.S.; Lovett, D.H.; Baker, A.J. Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc. Res. 2006, 69, 688–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashidani, S.; Tsutsui, H.; Ikeuchi, M.; Shiomi, T.; Matsusaka, H.; Kubota, T.; Imanaka-Yoshida, K.; Itoh, T.; Takeshita, A. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1229–H1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsman, P.; vant Hof, A.W.; de Boer, M.J.; Hoorntje, J.C.; Suryapranata, H.; Dambrink, J.H.; Zijlstra, F. Role of collateral circulation in the acute phase of ST-segment-elevation myocardial infarction treated with primary coronary intervention. Eur. Heart J. 2004, 25, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Sabia, P.J.; Powers, E.R.; Ragosta, M.; Sarembock, I.J.; Burwell, L.R.; Kaul, S. An association between collateral blood flow and myocardial viability in patients with recent myocardial infarction. N. Engl. J. Med. 1992, 327, 1825–1831. [Google Scholar] [CrossRef]
- Duvall, C.L.; Weiss, D.; Robinson, S.T.; Alameddine, F.M.; Guldberg, R.E.; Taylor, W.R. The role of osteopontin in recovery from hind limb ischemia. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyle, A.N.; Joseph, G.; Fan, A.E.; Weiss, D.; Landázuri, N.; Taylor, W.R. Reactive oxygen species regulate osteopontin expression in a murine model of postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1383–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef]
- Koshikawa, M.; Aizawa, K.; Kasai, H.; Izawa, A.; Tomita, T.; Kumazaki, S.; Tsutsui, H.; Koyama, J.; Shimodaira, S.; Takahashi, M.; et al. Elevated osteopontin levels in patients with peripheral arterial disease. Angiology 2009, 60, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Johnson, J.N.; Singh, K.; Singh, M. Impairment of myocardial angiogenic response in the absence of osteopontin. Microcirculation 2007, 14, 233–240. [Google Scholar] [CrossRef]
- Seo, K.W.; Lee, S.J.; Ye, B.H.; Kim, Y.W.; Bae, S.S.; Kim, C.D. Mechanical stretch enhances the expression and activity of osteopontin and MMP-2 via the Akt1/AP-1 pathways in VSMC. J. Mol. Cell. Cardiol. 2015, 85, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Zugck, C.; Nelles, M.; Juenger, C.; Frank, D.; Remppis, A.; Giannitsis, E.; Katus, H.A.; Frey, N. Osteopontin, a new prognostic biomarker in patients with chronic heart failure. Circ. Heart Fail. 2008, 1, 43–49. [Google Scholar] [CrossRef] [Green Version]
- López, B.; González, A.; Lindner, D.; Westermann, D.; Ravassa, S.; Beaumont, J.; Gallego, I.; Zudaire, A.; Brugnolaro, C.; Querejeta, R.; et al. Osteopontin-mediated myocardial fibrosis in heart failure: A role for lysyl oxidase? Cardiovasc. Res. 2013, 99, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francia, P.; Adduci, C.; Semprini, L.; Borro, M.; Ricotta, A.; Sensini, I.; Santini, D.; Caprinozzi, M.; Balla, C.; Simmaco, M.; et al. Osteopontin and galectin-3 predict the risk of ventricular tachycardia and fibrillation in heart failure patients with implantable defibrillators. J. Cardiovasc. Electrophysiol. 2014, 25, 609–616. [Google Scholar] [CrossRef]
- Satoh, M.; Nakamura, M.; Akatsu, T.; Shimoda, Y.; Segawa, I.; Hiramori, K. Myocardial osteopontin expression is associated with collagen fibrillogenesis in human dilated cardiomyopathy. Eur. J. Heart Fail. 2005, 7, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Cabiati, M.; Svezia, B.; Matteucci, M.; Botta, L.; Pucci, A.; Rinaldi, M.; Caselli, C.; Lionetti, V.; Del Ry, S. Myocardial Expression Analysis of Osteopontin and Its Splice Variants in Patients Affected by End-Stage Idiopathic or Ischemic Dilated Cardiomyopathy. PLoS ONE 2016, 11, e0160110. [Google Scholar] [CrossRef] [Green Version]
- Rubiś, P.; Wiśniowska-Śmiałek, S.; Dziewięcka, E.; Rudnicka-Sosin, L.; Kozanecki, A.; Podolec, P. Prognostic value of fibrosis-related markers in dilated cardiomyopathy: A link between osteopontin and cardiovascular events. Adv. Med. Sci. 2018, 63, 160–166. [Google Scholar] [CrossRef]
- Dai, J.; Matsui, T.; Abel, E.D.; Dedhar, S.; Gerszten, R.E.; Seidman, C.E.; Seidman, J.G.; Rosenzweig, A. Deep sequence analysis of gene expression identifies osteopontin as a downstream effector of integrin-linked kinase (ILK) in cardiac-specific ILK knockout mice. Circ. Heart Fail. 2014, 7, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wang, W.; Zhang, J.; Liang, T.; Fan, G.P.; Wang, Z.W.; Zhang, P.D.; Wang, X.; Zhang, J. Inhibition of osteopontin reduce the cardiac myofibrosis in dilated cardiomyopathy via focal adhesion kinase mediated signaling pathway. Am. J. Transl. Res. 2016, 8, 3645–3655. [Google Scholar]
- Ikeda, T.; Shirasawa, T.; Esaki, Y.; Yoshiki, S.; Hirokawa, K. Osteopontin mRNA is expressed by smooth muscle-derived foam cells in human atherosclerotic lesions of the aorta. J. Clin. Investig. 1993, 92, 2814–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strobescu-Ciobanu, C.; Giuşcă, S.E.; Căruntu, I.D.; Amălinei, C.; Rusu, A.; Cojocaru, E.; Popa, R.F.; Lupaşcu, C.D. Osteopontin and osteoprotegerin in atherosclerotic plaque—Are they significant markers of plaque vulnerability? Rom. J. Morphol. Embryol. 2020, 61, 793–801. [Google Scholar] [CrossRef]
- Mazzone, A.; Parri, M.S.; Giannessi, D.; Ravani, M.; Vaghetti, M.; Altieri, P.; Casalino, L.; Maltinti, M.; Balbi, M.; Barsotti, A.; et al. Osteopontin plasma levels and accelerated atherosclerosis in patients with CAD undergoing PCI: A prospective clinical study. Coron. Artery Dis. 2011, 22, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polonskaya, Y.V.; Kashtanova, E.V.; Murashov, I.S.; Kurguzov, A.V.; Sadovski, E.V.; Maslatsov, N.A.; Stakhneva, E.M.; Chernyavskii, A.M.; Ragino, Y.I. The Influence of Calcification Factors and Endothelial-Dysfunction Factors on the Development of Unstable Atherosclerotic Plaques. Diagnostics 2020, 10, 1074. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Rittling, S.R.; Okamoto, H.; Inobe, M.; Jia, N.; Shimizu, T.; Akino, M.; Sugawara, T.; Morimoto, J.; Kimura, C.; et al. Osteopontin deficiency attenuates atherosclerosis in female apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1029–1034. [Google Scholar] [CrossRef]
- Isoda, K.; Kamezawa, Y.; Ayaori, M.; Kusuhara, M.; Tada, N.; Ohsuzu, F. Osteopontin transgenic mice fed a high-cholesterol diet develop early fatty-streak lesions. Circulation 2003, 107, 679–681. [Google Scholar] [CrossRef] [Green Version]
- Kamińska, J.; Stopiński, M.; Mucha, K.; Pac, M.; Gołębiowski, M.; Niewczas, M.A.; Pączek, L.; Foroncewicz, B. Circulating Osteoprotegerin in Chronic Kidney Disease and All-Cause Mortality. Int. J. Gen. Med. 2021, 14, 2413–2420. [Google Scholar] [CrossRef]
- Speer, M.Y.; Chien, Y.C.; Quan, M.; Yang, H.Y.; Vali, H.; McKee, M.D.; Giachelli, C.M. Smooth muscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro. Cardiovasc. Res. 2005, 66, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Jono, S.; Peinado, C.; Giachelli, C.M. Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J. Biol. Chem. 2000, 275, 20197–20203. [Google Scholar] [CrossRef] [Green Version]
- Grau, J.B.; Poggio, P.; Sainger, R.; Vernick, W.J.; Seefried, W.F.; Branchetti, E.; Field, B.C.; Bavaria, J.E.; Acker, M.A.; Ferrari, G. Analysis of osteopontin levels for the identification of asymptomatic patients with calcific aortic valve disease. Ann. Thorac. Surg. 2012, 93, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muka, T.; Koromani, F.; Portilla, E.; O’Connor, A.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; Franco, O.H. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int. J. Cardiol. 2016, 212, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- Khyzha, N.; Alizada, A.; Wilson, M.D.; Fish, J.E. Epigenetics of Atherosclerosis: Emerging Mechanisms and Methods. Trends Mol. Med. 2017, 23, 332–347. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Q.; Li, D.; Li, J.; Aki, D.; Liu, Y.C. The E3 ligase VHL controls alveolar macrophage function via metabolic-epigenetic regulation. J. Exp. Med. 2018, 215, 3180–3193. [Google Scholar] [CrossRef]
- Chang, S.; Huang, J.; Niu, H.; Wang, J.; Si, Y.; Bai, Z.; Cheng, S.; Ding, W. Epigenetic regulation of osteopontin splicing isoform c defines its role as a microenvironmental factor to promote the survival of colon cancer cells from 5-FU treatment. Cancer Cell Int. 2020, 20, 452. [Google Scholar] [CrossRef]
- Moss, A.J.; Sim, A.M.; Adamson, P.D.; Seidman, M.A.; Andrews, J.P.M.; Doris, M.K.; Shah, A.S.V.; BouHaidar, R.; Alcaide-Corral, C.J.; Williams, M.C.; et al. Ex vivo (18)F-fluoride uptake and hydroxyapatite deposition in human coronary atherosclerosis. Sci. Rep. 2020, 10, 20172. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; McKee, M.D.; Liaw, L.; Almeida, M.; Yang, H.; Giachelli, C.M. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am. J. Pathol. 2002, 161, 2035–2046. [Google Scholar] [CrossRef] [Green Version]
- Ohri, R.; Tung, E.; Rajachar, R.; Giachelli, C.M. Mitigation of ectopic calcification in osteopontin-deficient mice by exogenous osteopontin. Calcif. Tissue Int. 2005, 76, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Ruan, C.C.; Ma, Y.; Tang, X.F.; Wu, Q.H.; Wang, J.G.; Zhu, D.L.; Gao, P.J. Osteopontin regulates macrophage activation and osteoclast formation in hypertensive patients with vascular calcification. Sci. Rep. 2017, 7, 40253. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Wang, Y.; Zhang, R.; Gao, Q.; Liang, X.; Gao, L.; Jiang, Z.; Qiao, R.; Han, D.; Zhang, Y.; et al. MRI/optical dual-modality imaging of vulnerable atherosclerotic plaque with an osteopontin-targeted probe based on Fe(3)O(4) nanoparticles. Biomaterials 2017, 112, 336–345. [Google Scholar] [CrossRef]
- Li, S.; Gou, T.; Wang, Q.; Chen, M.; Chen, Z.; Xu, M.; Wang, Y.; Han, D.; Cao, R.; Liu, J.; et al. Ultrasound/Optical Dual-Modality Imaging for Evaluation of Vulnerable Atherosclerotic Plaques with Osteopontin Targeted Nanoparticles. Macromol. Biosci. 2020, 20, e1900279. [Google Scholar] [CrossRef]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.W.; Kim, K.W.; Jang, M.Y.; Seok Jang, H.; Yun, T.J.; Lee, S.H.; et al. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef]
- Williams, J.W.; Zaitsev, K.; Kim, K.W.; Ivanov, S.; Saunders, B.T.; Schrank, P.R.; Kim, K.; Elvington, A.; Kim, S.H.; Tucker, C.G.; et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 2020, 21, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Renault, M.A.; Robbesyn, F.; Réant, P.; Douin, V.; Daret, D.; Allières, C.; Belloc, I.; Couffinhal, T.; Arnal, J.F.; Klingel, K.; et al. Osteopontin expression in cardiomyocytes induces dilated cardiomyopathy. Circ. Heart Fail. 2010, 3, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, I.A.; Mraiche, F. Targeting osteopontin, the silent partner of Na+/H+ exchanger isoform 1 in cardiac remodeling. J. Cell. Physiol. 2015, 230, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, S.; Givvimani, S.; Bhatnagar, S.; Qipshidze, N.; Tyagi, S.C.; Kumar, A. Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J. Immunol. 2011, 187, 2723–2731. [Google Scholar] [CrossRef] [Green Version]
- Caballero, E.P.; Santamaría, M.H.; Corral, R.S. Endogenous osteopontin induces myocardial CCL5 and MMP-2 activation that contributes to inflammation and cardiac remodeling in a mouse model of chronic Chagas heart disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 11–23. [Google Scholar] [CrossRef]
- Kristen, A.V.; Rosenberg, M.; Lindenmaier, D.; Merkle, C.; Steen, H.; Andre, F.; Schönland, S.O.; Schnabel, P.A.; Schuster, T.; Röcken, C.; et al. Osteopontin: A novel predictor of survival in patients with systemic light-chain amyloidosis. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2014, 21, 202–210. [Google Scholar] [CrossRef]
- Pei, H.; Zhang, H.; Tian, C.; Sun, X.; Qian, X.; Meng, Y.; Guo, X.; Chang, Q. Proliferative Vascular Smooth Muscle Cells Stimulate Extracellular Matrix Production via Osteopontin/p38 MAPK Signaling Pathway. Cardiology 2021, 1–10. [Google Scholar] [CrossRef]
- Xie, Y.; Sakatsume, M.; Nishi, S.; Narita, I.; Arakawa, M.; Gejyo, F. Expression, roles, receptors, and regulation of osteopontin in the kidney. Kidney Int. 2001, 60, 1645–1657. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, J.M.; Hafer, C.; Faulhaber-Walter, R.; Kümpers, P.; Kielstein, J.T.; Haller, H.; Fliser, D. Osteopontin predicts survival in critically ill patients with acute kidney injury. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2011, 26, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitland, S.; Nakstad, E.R.; Berg, J.P.; Trøseid, A.S.; Brusletto, B.S.; Brunborg, C.; Lundqvist, C.; Sunde, K. Urine β-2-Microglobulin, Osteopontin, and Trefoil Factor 3 May Early Predict Acute Kidney Injury and Outcome after Cardiac Arrest. Crit. Care Res. Pract. 2019, 2019, 4384796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.K.; Tian, P.X.; Wang, X.Z.; Xue, W.J.; Ding, X.M.; Zheng, J.; Ding, C.G.; Mao, T.C.; Duan, W.L.; Xi, M. Kidney injury molecule-1 and osteopontin: New markers for prediction of early kidney transplant rejection. Mol. Immunol. 2013, 54, 457–464. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Shek, K.; Wang, S.; Huang, X.; Lau, A.; Yin, Z.; Sun, H.; Liu, W.; Garcia, B.; Rittling, S.; et al. Osteopontin expressed in tubular epithelial cells regulates NK cell-mediated kidney ischemia reperfusion injury. J. Immunol. 2010, 185, 967–973. [Google Scholar] [CrossRef]
- Cen, C.; Aziz, M.; Yang, W.L.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Osteopontin Blockade Attenuates Renal Injury After Ischemia Reperfusion by Inhibiting NK Cell Infiltration. Shock 2017, 47, 52–60. [Google Scholar] [CrossRef]
- Persy, V.P.; Verhulst, A.; Ysebaert, D.K.; De Greef, K.E.; De Broe, M.E. Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int. 2003, 63, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Igarashi, M.; Hirata, A.; Tsuchiya, H.; Sugiyama, K.; Morita, Y.; Jimbu, Y.; Ohnuma, H.; Daimon, M.; Tominaga, M.; et al. Progression of diabetic nephropathy enhances the plasma osteopontin level in type 2 diabetic patients. Endocr. J. 2004, 51, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Sano, M.; Lu, L.; Wang, W.; Zhang, Q.; Zhang, R.; Wang, L.; Chen, Q.; Fukuda, K.; Shen, W. Plasma concentrations of osteopontin, but not thrombin-cleaved osteopontin, are associated with the presence and severity of nephropathy and coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2010, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Gordin, D.; Forsblom, C.; Panduru, N.M.; Thomas, M.C.; Bjerre, M.; Soro-Paavonen, A.; Tolonen, N.; Sandholm, N.; Flyvbjerg, A.; Harjutsalo, V.; et al. Osteopontin is a strong predictor of incipient diabetic nephropathy, cardiovascular disease, and all-cause mortality in patients with type 1 diabetes. Diabetes Care 2014, 37, 2593–2600. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Bompada, P.; Atac, D.; Laakso, M.; Groop, L.; De Marinis, Y. Epigenetic regulation of glucose-stimulated osteopontin (OPN) expression in diabetic kidney. Biochem. Biophys. Res. Commun. 2016, 469, 108–113. [Google Scholar] [CrossRef]
- Susztak, K.; Böttinger, E.; Novetsky, A.; Liang, D.; Zhu, Y.; Ciccone, E.; Wu, D.; Dunn, S.; McCue, P.; Sharma, K. Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes 2004, 53, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Wolak, T.; Kim, H.; Ren, Y.; Kim, J.; Vaziri, N.D.; Nicholas, S.B. Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int. 2009, 76, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Irita, J.; Okura, T.; Jotoku, M.; Nagao, T.; Enomoto, D.; Kurata, M.; Desilva, V.R.; Miyoshi, K.; Matsui, Y.; Uede, T.; et al. Osteopontin deficiency protects against aldosterone-induced inflammation, oxidative stress, and interstitial fibrosis in the kidney. Am. J. Physiol. Ren. Physiol. 2011, 301, F833–F844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolak, T. Osteopontin—A multi-modal marker and mediator in atherosclerotic vascular disease. Atherosclerosis 2014, 236, 327–337. [Google Scholar] [CrossRef]
- Lancha, A.; Rodríguez, A.; Catalán, V.; Becerril, S.; Sáinz, N.; Ramírez, B.; Burrell, M.A.; Salvador, J.; Frühbeck, G.; Gómez-Ambrosi, J. Osteopontin deletion prevents the development of obesity and hepatic steatosis via impaired adipose tissue matrix remodeling and reduced inflammation and fibrosis in adipose tissue and liver in mice. PLoS ONE 2014, 9, e98398. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, T.; Perez-Tilve, D.; Ogawa, D.; Gizard, F.; Zhao, Y.; Heywood, E.B.; Jones, K.L.; Kawamori, R.; Cassis, L.A.; Tschöp, M.H.; et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J. Clin. Investig. 2007, 117, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Endo, J.; Katsumata, Y.; Yamamoto, T.; Kataoka, M.; Isobe, S.; Yoshida, N.; Fukuda, K.; Sano, M. Negative legacy of obesity. PLoS ONE 2017, 12, e0186303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna-Luna, M.; Criales-Vera, S.; Medina-Leyte, D.; Díaz-Zamudio, M.; Flores-Zapata, A.; Cruz-Robles, D.; López-Meneses, M.; Olvera-Cruz, S.; Ramírez-Marroquín, S.; Flores-Castillo, C.; et al. Bone Morphogenetic Protein-2 and Osteopontin Gene Expression in Epicardial Adipose Tissue from Patients with Coronary Artery Disease Is Associated with the Presence of Calcified Atherosclerotic Plaques. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 1943–1951. [Google Scholar] [CrossRef]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Y.; Huang, D.; Luo, X.; Zhang, Y. Relationship of osteopontin and renal function with severity of coronary artery lesions. Int. J. Clin. Exp. Med. 2014, 7, 1122–1127. [Google Scholar]
- Lorenzen, J.; Krämer, R.; Kliem, V.; Bode-Boeger, S.M.; Veldink, H.; Haller, H.; Fliser, D.; Kielstein, J.T. Circulating levels of osteopontin are closely related to glomerular filtration rate and cardiovascular risk markers in patients with chronic kidney disease. Eur. J. Clin. Investig. 2010, 40, 294–300. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Rådholm, K.; Figtree, G.; Perkovic, V.; Solomon, S.D.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Barrett, T.D.; Shaw, W.; Desai, M.; et al. Canagliflozin and Heart Failure in Type 2 Diabetes Mellitus: Results From the CANVAS Program. Circulation 2018, 138, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef] [Green Version]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzén, S.; Palm, F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Ren. Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef] [Green Version]
- Sharma, I.; Deng, F.; Liao, Y.; Kanwar, Y.S. Myo-inositol Oxygenase (MIOX) Overexpression Drives the Progression of Renal Tubulointerstitial Injury in Diabetes. Diabetes 2020, 69, 1248–1263. [Google Scholar] [CrossRef]
- Fleury, C.; Mignotte, B.; Vayssière, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef]
- Sano, M.; Goto, S. Possible Mechanism of Hematocrit Elevation by Sodium Glucose Cotransporter 2 Inhibitors and Associated Beneficial Renal and Cardiovascular Effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirakawa, K.; Sano, M. Osteopontin in Cardiovascular Diseases. Biomolecules 2021, 11, 1047. https://doi.org/10.3390/biom11071047
Shirakawa K, Sano M. Osteopontin in Cardiovascular Diseases. Biomolecules. 2021; 11(7):1047. https://doi.org/10.3390/biom11071047
Chicago/Turabian StyleShirakawa, Kohsuke, and Motoaki Sano. 2021. "Osteopontin in Cardiovascular Diseases" Biomolecules 11, no. 7: 1047. https://doi.org/10.3390/biom11071047
APA StyleShirakawa, K., & Sano, M. (2021). Osteopontin in Cardiovascular Diseases. Biomolecules, 11(7), 1047. https://doi.org/10.3390/biom11071047