
Ceramide Metabolism and Parkinson’s Disease—Therapeutic Targets

 , ,
, ,  and
and 
Abstract
:1. Introduction
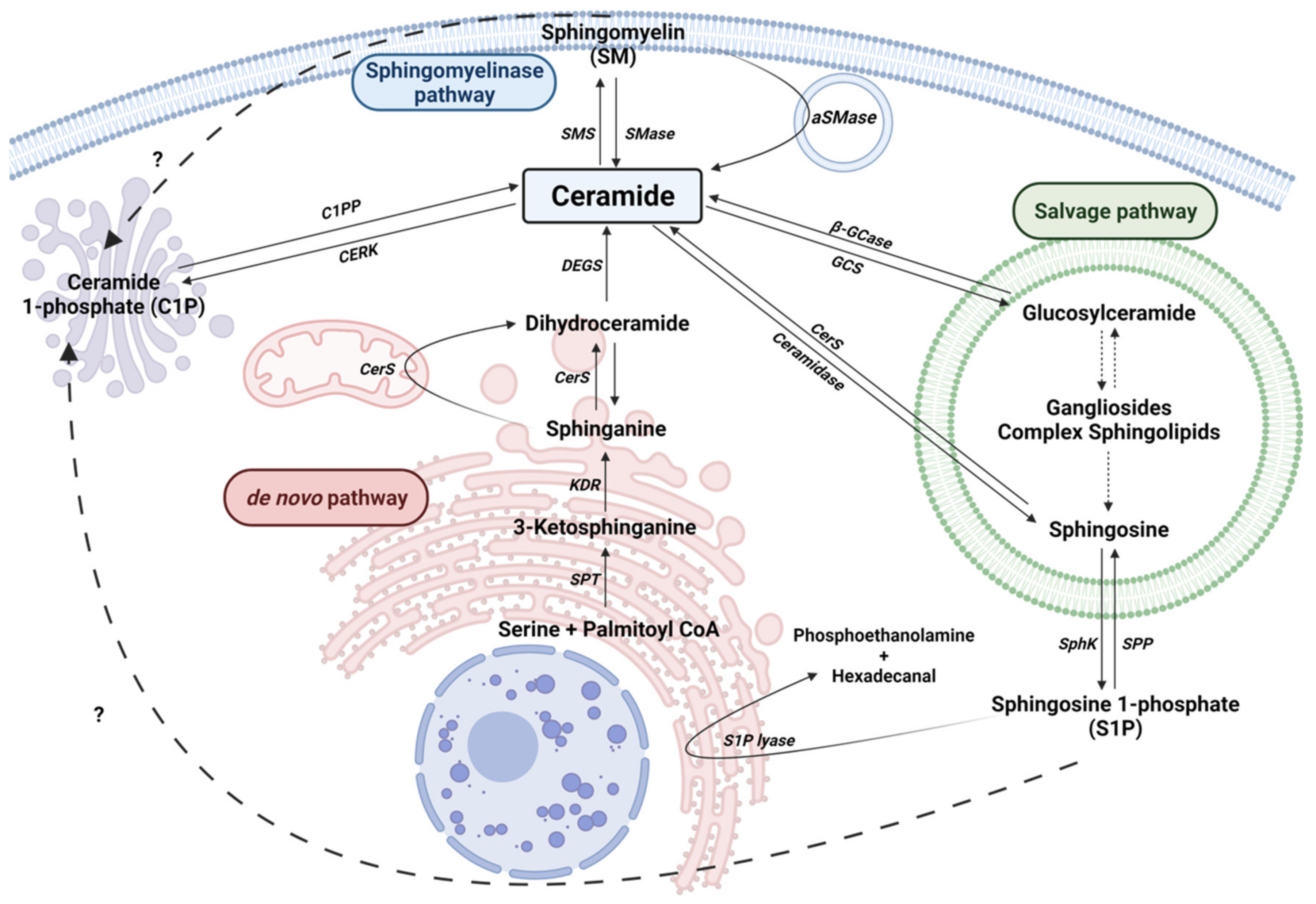
2. Sphingolipid Metabolism
2.1. The de novo Pathway
2.2. The Sphingomyelinase (SMase) Pathway
2.3. The Salvage Pathway
2.4. Ceramide Kinase/Ceramide 1-Phosphate Phosphatase (CerK/CPP) and Sphingosine Kinase/Sphingosine 1-Phosphate Phosphatase (SphK/SPP) Axis
3. Neurodegeneration and Sphingolipid Metabolism
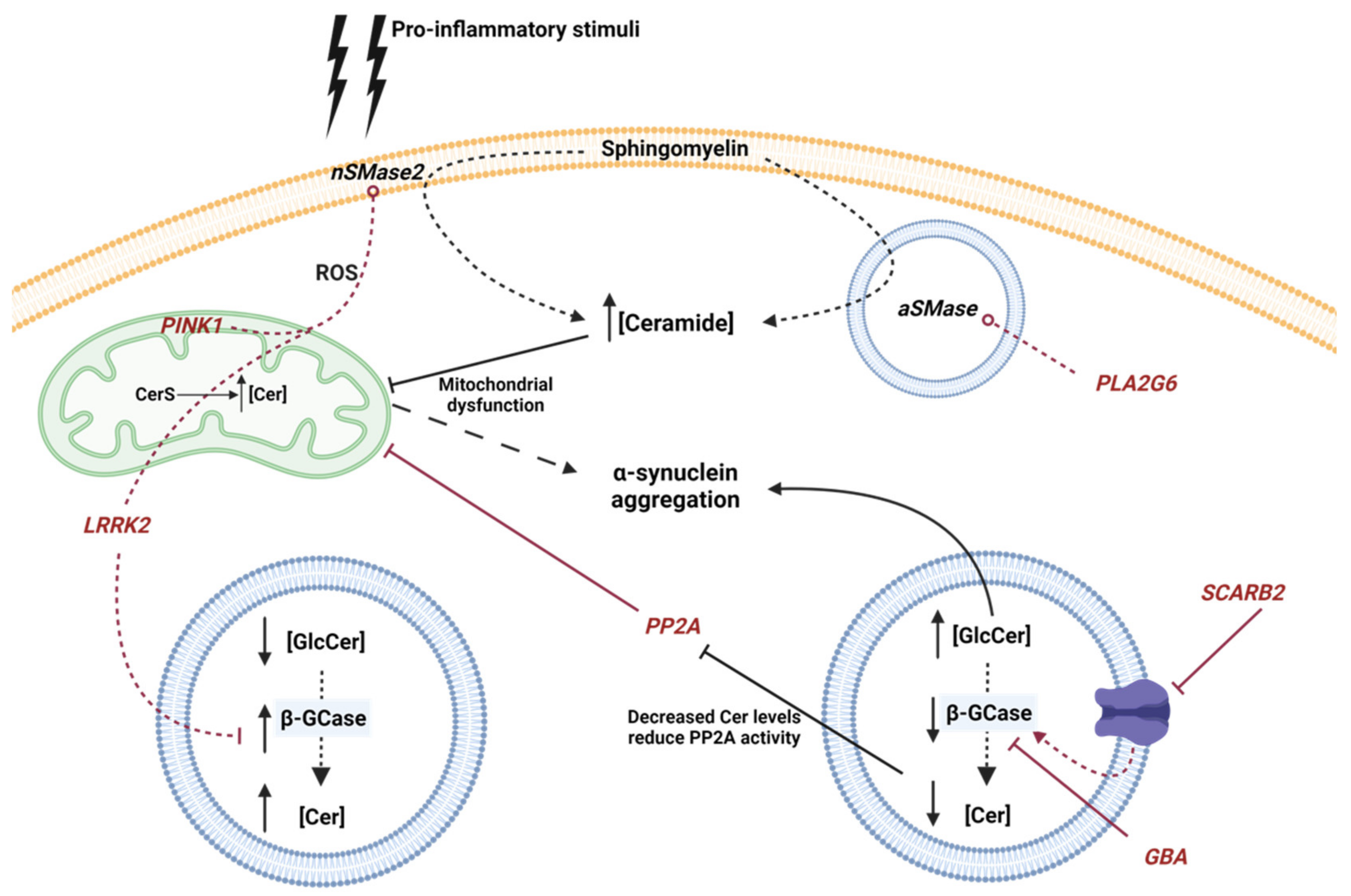
4. Ceramide Metabolism Alterations in Parkinson’s Disease
4.1. Genetic Risks
4.1.1. GBA
4.1.2. LRRK2
4.1.3. PLA2G6
4.1.4. PINK1
4.1.5. SCARB2
4.2. Enviromental Risks
5. Human Sphingolipidomics of Ceramide Metabolism in PD

{kind=link}
{kind=link}
| Sphingolipid Species | Levels | Source | Type of PD | Notes | Ref. |
|---|---|---|---|---|---|
| Cer | ↑ | Serum | GBA mutation | Possible early development of PD and worsening of symptoms | [164] |
| ↑ | Post-mortem CSF | Not specified | Correlation with neuropathological staging and disease duration | [202] | |
| ↑ | Primary visual cortex | Sporadic | Contribution to neuronal dysfunction | [205] | |
| Long-chain Cer (such as C24:1 and C24:0-Cer) | ↓ | Anterior cingulate cortex | Impartment of salvage pathway by increased CerS1 expression | [201] | |
| C14:0-Cer | ↑ | Plasma | PD with dementia | Association with delayed free recall and cognition | [203] |
| C16:0-Cer | ↑ | Plasma | Non-GBA mutation | Association with worse cognition | [204] |
| C18:0-Cer | ↑ | Plasma | Non-GBA mutation | Association with worse cognition | [204] |
| ↑ * | PD with dementia | Association with sleep behaviour disturbance | [203] | ||
| C18:1-Cer | ↓ | Frontal cortex | Not specified | Increased formation of diacylglycerols (DAGs) | [210] |
| C20:0-Cer | ↑ * | Plasma | PD with dementia | Association with anxiety | [203] |
| ↑ | Non-GBA mutation | Association with worse cognition | [204] | ||
| C22:0-Cer | ↑ | Plasma | Non-GBA mutation | Association with worse cognition | [204] |
| ↑ * | PD with dementia | Association with hallucination | [203] | ||
| C24:1-Cer | ↑ | Plasma | PD with dementia | Association with the score of immediate verbal recall and delayed free recall | [203] |
| ↑ | Non-GBA mutation | Association with worse cognition | [204] | ||
| GlcCer | ↑ | Plasma | Non-GBA or Non-LRRK2 G2019S mutation | Association with worse cognition | [204,218] |
| GlcCer C16:0 | ↑ | Plasma | Non-GBA mutation | Association with worse cognition | [204] |
| GlcCer C18:0 | ↑ * | Temporal cortex | Not specified | Correlation with PD severity | [209] |
| GlcCer C20:0 | ↑ * | Temporal cortex | Not specified | Correlation with PD severity | [209] |
| GlcCer C22:0 | ↑ * | Temporal cortex | Not specified | Correlation with PD severity | [209] |
| ↑ | Plasma | Non-GBA mutation | Tendency to association with worse cognition | [204] | |
| GlcCer C24:0 | ↑ | Plasma | Non-GBA mutation | Association with worse cognition | [204] |
| ↑ * | Temporal cortex | Not specified | Correlation with PD severity | [209] | |
| GlcCer C24:1 | ↑ * | Temporal cortex | Not specified | Correlation with PD severity | [209] |
| ↓ | Frontal cortex | Not specified | Increased formation of DAGs | [210] | |
| LacCer | ↑ | Serum | GBA mutation | Proposed as novel biomarker for increased risk of PD develop | [164] |
| C1P | ↓ | Serum | GBA mutation | Proposed as novel biomarker for increased risk of PD develop | [164] |
| SM | ↑ | Post-mortem CSF | Not specified | Correlation with neuropathological staging and disease duration | [205] |
| ↑ | Substantia nigra | Male PD | Caused by enrichment in Lewy bodies | [206] | |
| Long-chain SM | ↑ | Primary visual cortex | Sporadic | Contribution to neuronal dysfunction | [205] |
| SM d30:1 | ↓ * | Plasma | Not specified | Due to dysregulation of sphingolipids metabolism. Possibly involved in demyelination | [208] |
| SM d32:1 | ↓ * | Plasma | Not specified | Due to dysregulation of sphingolipids metabolism. Possibly involved in demyelination | [208] |
| SM d39:1 | ↓ * | Plasma | Not specified | Due to dysregulation of sphingolipids metabolism. Possibly involved in demyelination | [208] |
| GM1 | ↓ | Substantia nigra dopaminergic neurons | Sporadic | Loss of neuroprotection and acceleration of α-syn formation | [216] |
| Ganglioside-NANA-3 | ↑ | Plasma | GBA or/and LRRK2 G2019S mutation | Acceleration of α-syn formation | [217] |
| GM3 d18:1/24:1 | ↑ | Plasma | Non-GBA or Non-LRRK2 G2019S mutation | Acceleration of α-syn formation | [218] |
| GM3 d18:1/26:0 | ↑ | Plasma | Non-GBA or Non-LRRK2 G2019S mutation | Acceleration of α-syn formation | [218] |
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Dextera, D.T.; Jenner, P. Parkinson Disease: From Pathology to Molecular Disease Mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Von Campenhausen, S.; Winter, Y.; e Silva, A.R.; Sampaio, C.; Ruzicka, E.; Barone, P.; Poewe, W.; Guekht, A.; Mateus, C.; Pfeiffer, K.-P.; et al. Costs of Illness and Care in Parkinson’s Disease: An Evaluation in Six Countries. Eur. Neuropsychopharmacol. 2011, 21, 180–191. [Google Scholar] [CrossRef]
- Indellicato, R.; Trinchera, M. The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism. Int. J. Mol. Sci. 2019, 20, 3304. [Google Scholar] [CrossRef] [Green Version]
- Alessenko, A.V.; Albi, E. Exploring Sphingolipid Implications in Neurodegeneration. Front. Neurol. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Hussain, M.M. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends Endocrinol. Metab. 2017, 28, 506–518. [Google Scholar] [CrossRef]
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Role of Bioactive Sphingolipids in Physiology and Pathology. Essays Biochem. 2020, 64, 579–589. [Google Scholar] [CrossRef]
- Giussani, P.; Prinetti, A.; Tringali, C. The Role of Sphingolipids in Myelination and Myelin Stability and Their Involvement in Childhood and Adult Demyelinating Disorders. J. Neurochem. 2021, 156, 403–414. [Google Scholar] [CrossRef]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef]
- Ouro, A.; Arana, L.; Gangoiti, P.; Gomez-Muñoz, A. Role of Ceramide 1-Phosphate in the Regulation of Cell Survival and Inflammation. Biochemistry 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gomez-Munoz, A.; Gómez-Muñoz, A. Ceramide and Ceramide 1-Phosphate in Health and Disease. Lipids Health Dis. 2010, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gangoiti, P.; Camacho, L.; Arana, L.; Ouro, A.; Granado, M.H.; Brizuela, L.; Casas, J.; Fabrias, G.; Abad, J.L.; Delgado, A.; et al. Control of Metabolism and Signaling of Simple Bioactive Sphingolipids: Implications in Disease. Prog. Lipid Res. 2010, 49, 316–334. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.R.; Zetterberg, H.; Blennow, K.; Andreasson, U.; Johnson, S.C.; Gleason, C.E.; Blazel, H.M.; Puglielli, L.; et al. Cerebrospinal Fluid Sphingolipids, β-Amyloid, and Tau in Adults at Risk for Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 2486–2494. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Wang, L.; Marcogliese, P.C.; Bellen, H.J. Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends Endocrinol. Metab. 2019, 30, 106–117. [Google Scholar] [CrossRef]
- Van Kruining, D.; Luo, Q.; van Echten-Deckert, G.; Mielke, M.M.; Bowman, A.; Ellis, S.; Oliveira, T.G.; Martinez-Martinez, P. Sphingolipids as Prognostic Biomarkers of Neurodegeneration, Neuroinflammation, and Psychiatric Diseases and Their Emerging Role in Lipidomic Investigation Methods. Adv. Drug Deliv. Rev. 2020, 159, 232–244. [Google Scholar] [CrossRef]
- Gulbins, A.; Grassm, H.; Hoehn, R.; Wilker, B.; Soddemann, M.; Kohnen, M.; Edwards, M.J.; Kornhuber, J.; Gulbins, E. Regulation of Neuronal Stem Cell Proliferation in the Hippocampus by Endothelial Ceramide. Cell. Physiol. Biochem. 2016, 39, 790–801. [Google Scholar] [CrossRef]
- Schultz, A.; Larsson, C. Ceramide Influences Neurite Outgrowth and Neuroblastoma Cell Apoptosis Regulated by Novel Protein Kinase C Isoforms. J. Neurochem. 2004, 89, 1427–1435. [Google Scholar] [CrossRef]
- Cruciani-Guglielmacci, C.; López, M.; Campana, M.; le Stunff, H. Brain Ceramide Metabolism in the Control of Energy Balance. Front. Physiol. 2017, 8, 787. [Google Scholar] [CrossRef] [Green Version]
- Jana, A.; Hogan, E.L.; Pahan, K. Ceramide and Neurodegeneration: Susceptibility of Neurons and Oligodendrocytes to Cell Damage and Death. J. Neurol. Sci. 2009, 278, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Pujol-Lereis, L.M. Alteration of Sphingolipids in Biofluids: Implications for Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3564. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M. Sphingolipid Lysosomal Storage Disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef]
- Watters, R.J.; Kester, M.; Tran, M.A.; Loughran, T.P.; Liu, X. Development and Use of Ceramide Nanoliposomes in Cancer. Methods Enzymol. 2012, 508, 89–108. [Google Scholar] [CrossRef]
- Gomez-Muñoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordoñez, M. Control of Inflammatory Responses by Ceramide, Sphingosine 1-Phosphate and Ceramide 1-Phosphate. Prog. Lipid Res. 2016, 61, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Albeituni, S.; Stiban, J. Roles of Ceramides and Other Sphingolipids in Immune Cell Function and Inflammation. Adv. Exp. Med. Biol. 2019, 1161, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, B.W. Kicking off Sphingolipid Biosynthesis: Structures of the Serine Palmitoyltransferase Complex. Nat. Struct. Mol. Biol. 2021, 28, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.L.; Mestre, B.; Shin, S.-H.; Futerman, A.H. Ceramide Synthases: Reflections on the Impact of Dr. Lina M. Obeid. Cell. Signal. 2021, 82, 109958. [Google Scholar] [CrossRef]
- Mignard, V.; Dubois, N.; Lanoé, D.; Joalland, M.P.; Oliver, L.; Pecqueur, C.; Heymann, D.; Paris, F.; Vallette, F.M.; Lalier, L. Sphingolipids Distribution at Mitochondria-Associated Membranes (MAM) upon Induction of Apoptosis. J. Lipid Res. 2020, 61, 1025–1037. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Gudz, T.I. Ceramide and Mitochondria in Ischemic Brain Injury. Int. J. Biochem. Mol. Biol. 2011, 2, 347–361. [Google Scholar]
- Yu, J.; Novgorodov, S.A.; Chudakova, D.; Zhu, H.; Bielawska, A.; Bielawski, J.; Obeid, L.M.; Kindy, M.S.; Gudz, T.I. JNK3 Signaling Pathway Activates Ceramide Synthase Leading to Mitochondrial Dysfunction. J. Biol. Chem. 2007, 282, 25940–25949. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, B.; Tippetts, T.S.; Monibas, R.M.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a Ceramide Double Bond Improves Insulin Resistance and Hepatic Steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef]
- Ohi, K.; Ursini, G.; Li, M.; Shin, J.H.; Ye, T.; Chen, Q.; Tao, R.; Kleinman, J.E.; Hyde, T.M.; Hashimoto, R.; et al. DEGS2 Polymorphism Associated with Cognition in Schizophrenia Is Associated with Gene Expression in Brain. Transl. Psychiatry 2015, 5, e550. [Google Scholar] [CrossRef] [Green Version]
- Karsai, G.; Kraft, F.; Haag, N.; Korenke, G.C.; Hänisch, B.; Othman, A.; Suriyanarayanan, S.; Steiner, R.; Knopp, C.; Mull, M.; et al. DEGS1-Associated Aberrant Sphingolipid Metabolism Impairs Nervous System Function in Humans. J. Clin. Investig. 2019, 129, 1229–1239. [Google Scholar] [CrossRef] [Green Version]
- Goi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and Membrane Activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, A.; Illes, K.; Heinz, L.X.; Superti-Furga, G.; Nagar, B. Crystal Structure of Mammalian Acid Sphingomyelinase. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Clarke, C.J.; Snook, C.F.; Tani, M.; Matmati, N.; Marchesini, N.; Hannun, Y.A. The Extended Family of Neutral Sphingomyelinases. Biochemistry 2006, 45, 11247–11256. [Google Scholar] [CrossRef]
- Cataldi, S.; Borrelli, A.; Ceccarini, M.R.; Nakashidze, I.; Codini, M.; Belov, O.; Ivanov, A.; Krasavin, E.; Ferri, I.; Conte, C.; et al. Acid and Neutral Sphingomyelinase Behavior in Radiation-Induced Liver Pyroptosis and in the Protective/Preventive Role of RMnSOD. Int. J. Mol. Sci. 2020, 21, 3281. [Google Scholar] [CrossRef]
- Kornhuber, J.; Rhein, C.; Müller, C.P.; Mühle, C. Secretory Sphingomyelinase in Health and Disease. Biol. Chem. 2015, 396, 707–736. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Canals, D.; Idkowiak-Baldys, J.; Simbari, F.; Roddy, P.; Perry, D.M.; Kitatani, K.; Luberto, C.; Hannun, Y.A. Regulated Secretion of Acid Sphingomyelinase: Implications for Selectivity of Ceramide Formation. J. Biol. Chem. 2010, 285, 35706–35718. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.A.; Riethmuller, J.; Luth, A.; Doring, G.; Kleuser, B.; Gulbins, E. Acid Sphingomyelinase Inhibitors Normalize Pulmonary Ceramide and Inflammation in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 716–724. [Google Scholar] [CrossRef]
- Gomez-Muñoz, A.; Gangoiti, P.; Arana, L.; Ouro, A.; Rivera, I.G.; Ordoñez, M.; Trueba, M. New Insights on the Role of Ceramide 1-Phosphate in Inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 1060–1066. [Google Scholar] [CrossRef]
- Malaplate-Armand, C.; Florent-Béchard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble Oligomers of Amyloid-β Peptide Induce Neuronal Apoptosis by Activating a CPLA2-Dependent Sphingomyelinase-Ceramide Pathway. Neurobiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef]
- Morad, S.A.F.; Cabot, M.C. Ceramide-Orchestrated Signalling in Cancer Cells. Nat. Rev. Cancer 2012, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, K.; Erlich, S.; Perl, D.P.; Ferlinz, K.; Bisgaier, C.L.; Sandhoff, K.; Desnick, R.J.; Stewart, C.L.; Schuchman, E.H. Acid Sphingomyelinase Deficient Mice: A Model of Types A and B Niemann–Pick Disease. Nat. Genet. 1995, 10, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Wasserstein, M.P. Types A and B Niemann-Pick Disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.X.; Clarke, C.J.; Hannun, Y.A. Mammalian Neutral Sphingomyelinases: Regulation and Roles in Cell Signaling Responses. NeuroMolecular Med. 2010, 12, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Shamseddine, A.A.; Airola, M.V.; Hannun, Y.A. Roles and Regulation of Neutral Sphingomyelinase-2 in Cellular and Pathological Processes. Adv. Biol. Regul. 2015, 57, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and Characterization of the Mammalian Brain-Specific, Mg2+-Dependent Neutral Sphingomyelinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5895–5900. [Google Scholar] [CrossRef] [Green Version]
- Cataldi, S.; Arcuri, C.; Hunot, S.; Légeron, F.P.; Mecca, C.; Garcia-Gil, M.; Lazzarini, A.; Codini, M.; Beccari, T.; Tasegian, A.; et al. Neutral Sphingomyelinase Behaviour in Hippocampus Neuroinflammation of MPTP-Induced Mouse Model of Parkinson’s Disease and in Embryonic Hippocampal Cells. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Tabatadze, N.; Savonenko, A.; Song, H.; Bandaru, V.V.R.; Chu, M.; Haughey, N.J. Inhibition of Neutral Sphingomyelinase-2 Perturbs Brain Sphingolipid Balance and Spatial Memory in Mice. J. Neurosci. Res. 2010, 88, 2940–2951. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.Z.; Huang, B.S.; Shen, W.; Gao, L.; Ding, Z.Z.; Wu, H.W.; Guo, J. Early Activation of NSMase2/Ceramide Pathway in Astrocytes Is Involved in Ischemia-Associated Neuronal Damage via Inflammation in Rat Hippocampi. J. Neuroinflamm. 2013, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher Disease: Mutation and Polymorphism Spectrum in the Glucocerebrosidase Gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Velayati, A.; Yu, W.H.; Sidransky, E. The Role of Glucocerebrosidase Mutations in Parkinson Disease and Lewy Body Disorders. Curr. Neurol. Neurosci. Rep. 2010, 10, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Coant, N.; Hannun, Y.A. Neutral Ceramidase: Advances in Mechanisms, Cell Regulation, and Roles in Cancer. Adv. Biol. Regul. 2019, 71, 141–146. [Google Scholar] [CrossRef]
- Romiti, E.; Meacci, E.; Tani, M.; Nuti, F.; Farnararo, M.; Ito, M.; Bruni, P. Neutral/Alkaline and Acid Ceramidase Activities Are Actively Released by Murine Endothelial Cells. Biochem. Biophys. Res. Commun. 2000, 275, 746–751. [Google Scholar] [CrossRef]
- Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A.; Gomez-Muñoz, A.; Gomez-Munoz, A. Activation of Protein Kinase C-Alpha Is Essential for Stimulation of Cell Proliferation by Ceramide 1-Phosphate. FEBS Lett. 2010, 584, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Gangoiti, P.; Bernacchioni, C.; Donati, C.; Cencetti, F.; Ouro, A.; Gómez-Muñoz, A.; Bruni, P.; Gomez-Munoz, A.; Bruni, P. Ceramide 1-Phosphate Stimulates Proliferation of C2C12 Myoblasts. Biochimie 2012, 94, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Ouro, A.; Arana, L.; Gangoiti, P.; Rivera, I.G.; Ordoñez, M.; Trueba, M.; Lankalapalli, R.S.; Bittman, R.; Gomez-Muñoz, A. Ceramide 1-Phosphate Stimulates Glucose Uptake in Macrophages. Cell. Signal. 2013, 25, 786–795. [Google Scholar] [CrossRef] [Green Version]
- Gangoiti, P.; Granado, M.H.; Wei, S.; Kong, J.Y.; Steinbrecher, U.P.; Gómez-muñoz, A. Ceramide 1-Phosphate Stimulates Macrophage Proliferation through Activation of the PI3-Kinase / PKB, JNK and ERK1 / 2 Pathways. Cell. Signal. 2008, 20, 726–736. [Google Scholar] [CrossRef]
- Ouro, A.; Arana, L.; Riazy, M.; Zhang, P.; Gomez-Larrauri, A.; Steinbrecher, U.; Duronio, V.; Gomez-Muñoz, A. Vascular Endothelial Growth Factor Mediates Ceramide 1-Phosphate-Stimulated Macrophage Proliferation. Exp. Cell Res. 2017, 361, 277–283. [Google Scholar] [CrossRef]
- Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A.; Gómez-Muñoz, A. Involvement of Nitric Oxide in the Promotion of Cell Survival by Ceramide 1-Phosphate. FEBS Lett. 2008, 582, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Munoz, A.; Kong, J.; Salh, B.; Steinbrecher, U.P. Sphingosine-1-Phosphate Inhibits Acid Sphingomyelinase and Blocks Apoptosis in Macrophages. FEBS Lett. 2003, 539, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Newcomb, B.; Rhein, C.; Mileva, I.; Ahmad, R.; Clarke, C.J.; Snider, J.; Obeid, L.M.; Hannun, Y.A. Identification of an Acid Sphingomyelinase Ceramide Kinase Pathway in the Regulation of the Chemokine CCL5. J. Lipid Res. 2018, 59, 1219–1229. [Google Scholar] [CrossRef] [Green Version]
- Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; Gómez-Muñoz, A. Ceramide 1-Phosphate Inhibits Serine Palmitoyltransferase and Blocks Apoptosis in Alveolar Macrophages. Biochim. Biophys. Acta 2009, 1791, 263–272. [Google Scholar] [CrossRef]
- Gomez-Munoz, A.; Kong, J.Y.; Parhar, K.; Wang, S.W.; Gangoiti, P.; Gonzalez, M.; Eivemark, S.; Salh, B.; Duronio, V.; Steinbrecher, U.P. Ceramide-1-Phosphate Promotes Cell Survival through Activation of the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. FEBS Lett. 2005, 579, 3744–3750. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.K.; Gao, Y.G.; Deng, Y.; Chalfant, C.E.; Hinchcliffe, E.H.; Brown, R.E. CPTP: A Sphingolipid Transfer Protein That Regulates Autophagy and Inflammasome Activation†. Autophagy 2018, 14, 862–879. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Wang, W.; McGiffert, C.; Huang, M.C.; Graler, M.H. Sphingosine 1-Phosphate and Its G Protein-Coupled Receptors Constitute a Multifunctional Immunoregulatory System. J. Cell Biochem. 2004, 92, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Gaire, B.P.; Choi, J.W. Sphingosine 1-Phosphate Receptors in Cerebral Ischemia. NeuroMol. Med. 2021, 23, 211–223. [Google Scholar] [CrossRef]
- Calise, S.; Blescia, S.; Cencetti, F.; Bernacchioni, C.; Donati, C.; Bruni, P. Sphingosine 1-Phosphate Stimulates Proliferation and Migration of Satellite Cells: Role of S1P Receptors. Biochim. Biophys. Acta 2012, 1823, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Cartier, A.; Leigh, T.; Liu, C.H.; Hla, T. Endothelial Sphingosine 1-Phosphate Receptors Promote Vascular Normalization and Antitumor Therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3157–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, J.D. Fifty Years of Lyase and a Moment of Truth: Sphingosine Phosphate Lyase from Discovery to Disease. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitroi, D.N.; Karunakaran, I.; Gräler, M.; Saba, J.D.; Ehninger, D.; Ledesma, M.D.; van Echten-Deckert, G. SGPL1 (Sphingosine Phosphate Lyase 1) Modulates Neuronal Autophagy via Phosphatidylethanolamine Production. Autophagy 2017, 13, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and Basic Pathology of Alzheimer Disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Bandaru, V.V.R.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B. Serum Ceramides Increase the Risk of Alzheimer Disease: The Women’s Health and Aging Study II. Neurology 2012, 79, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased Ceramide in Brains with Alzheimer’s and Other Neurodegenerative Diseases. J. Alzheimer’s Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Czubowicz, K.; Jęśko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2019, 56, 5436–5455. [Google Scholar] [CrossRef] [Green Version]
- Panchal, M.; Gaudin, M.; Lazar, A.N.; Salvati, E.; Rivals, I.; Ayciriex, S.; Dauphinot, L.; Dargère, D.; Auzeil, N.; Masserini, M.; et al. Ceramides and Sphingomyelinases in Senile Plaques. Neurobiol. Dis. 2014, 65, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Ellis, B.C.; Saunders, A.J.; Kovacs, D.M. Ceramide Stabilizes β-Site Amyloid Precursor Protein-Cleaving Enzyme 1 and Promotes Amyloid β-Peptide Biogenesis. J. Biol. Chem. 2003, 278, 19777–19783. [Google Scholar] [CrossRef] [Green Version]
- Desbène, C.; Malaplate-Armand, C.; Youssef, I.; Garcia, P.; Stenger, C.; Sauvée, M.; Fischer, N.; Rimet, D.; Koziel, V.; Escanyé, M.-C.; et al. Critical Role of CPLA2 in Aβ Oligomer-Induced Neurodegeneration and Memory Deficit. Neurobiol. Aging 2012, 33, 1123.e17–1123.e29. [Google Scholar] [CrossRef]
- Jana, A.; Pahan, K. Fibrillar Amyloid-β-Activated Human Astroglia Kill Primary Human Neurons via Neutral Sphingomyelinase: Implications for Alzheimer’s Disease. J. Neurosci. 2010, 30, 12676–12689. [Google Scholar] [CrossRef]
- Lee, J.T.; Xu, J.; Lee, J.M.; Ku, G.; Han, X.; Yang, D.I.; Chen, S.; Hsu, C.Y. Amyloid-β Peptide Induces Oligodendrocyte Death by Activating the Neutral Sphingomyelinase-Ceramide Pathway. J. Cell Biol. 2004, 164, 123–131. [Google Scholar] [CrossRef]
- Yang, D.I.; Yeh, C.H.; Chen, S.; Xu, J.; Hsu, C.Y. Neutral Sphingomyelinase Activation in Endothelial and Glial Cell Death Induced by Amyloid Beta-Peptide. Neurobiol. Dis. 2004, 17, 99–107. [Google Scholar] [CrossRef]
- Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; et al. BACE1 Activity Is Modulated by Cell-Associated Sphingosine-1-Phosphate. J. Neurosci. 2011, 31, 6850–6857. [Google Scholar] [CrossRef] [Green Version]
- Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C.; et al. Reduced Sphingosine Kinase-1 and Enhanced Sphingosine 1-Phosphate Lyase Expression Demonstrate Deregulated Sphingosine 1-Phosphate Signaling in Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 1–10. [Google Scholar] [CrossRef]
- Paciotti, S.; Albi, E.; Parnetti, L.; Beccari, T. Lysosomal Ceramide Metabolism Disorders: Implications in Parkinson’s Disease. J. Clin. Med. 2020, 9, 594. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Ren, J.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Lysosome Sorting of β-Glucocerebrosidase by LIMP-2 Is Targeted by the Mannose 6-Phosphate Receptor. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. A Rare Lysosomal Enzyme Gene SMPD1 Variant (p.R591C) Associates with Parkinson’s Disease. Neurobiol. Aging 2013, 34, 2890.e13–2890.e15. [Google Scholar] [CrossRef]
- Conte, C.; Arcuri, C.; Cataldi, S.; Mecca, C.; Codini, M.; Ceccarini, M.R.; Patria, F.F.; Beccari, T.; Albi, E. Niemann-Pick Type a Disease: Behavior of Neutral Sphingomyelinase and Vitamin D Receptor. Int. J. Mol. Sci. 2019, 20, 2365. [Google Scholar] [CrossRef] [Green Version]
- Vanier, M.T. Niemann-Pick diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 113, pp. 1717–1721. [Google Scholar]
- Torres, S.; Solsona-Vilarrasa, E.; Nuñez, S.; Matías, N.; Insausti-Urkia, N.; Castro, F.; Casasempere, M.; Fabriás, G.; Casas, J.; Enrich, C.; et al. Acid Ceramidase Improves Mitochondrial Function and Oxidative Stress in Niemann-Pick Type C Disease by Repressing STARD1 Expression and Mitochondrial Cholesterol Accumulation. Redox Biol. 2021, 102052. [Google Scholar] [CrossRef]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine Accumulation in Tissues from Patients with Gaucher Disease: Correlation with Phenotype and Genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid Ceramidase Deficiency: Farber Disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozma, C.; Iurașcu, M.-I.; Eichler, S.; Hovakimyan, M.; Brandau, O.; Zielke, S.; Böttcher, T.; Giese, A.-K.; Lukas, J.; Rolfs, A. C26-Ceramide as Highly Sensitive Biomarker for the Diagnosis of Farber Disease. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Spratley, S.J.; Hill, C.H.; Viuff, A.H.; Edgar, J.R.; Skjødt, K.; Deane, J.E. Molecular Mechanisms of Disease Pathogenesis Differ in Krabbe Disease Variants. Traffic 2016, 17, 908–922. [Google Scholar] [CrossRef]
- Marshall, M.S.; Bongarzone, E.R. Beyond Krabbe’s Disease: The Potential Contribution of Galactosylceramidase Deficiency to Neuronal Vulnerability in Late-Onset Synucleinopathies. J. Neurosci. Res. 2016, 94, 1328–1332. [Google Scholar] [CrossRef] [Green Version]
- Maglione, V.; Marchi, P.; di Pardo, A.; Lingrell, S.; Horkey, M.; Tidmarsh, E.; Sipione, S. Impaired Ganglioside Metabolism in Huntington’s Disease and Neuroprotective Role of GM1. J. Neurosci. 2010, 30, 4072–4080. [Google Scholar] [CrossRef]
- Alpaugh, M.; Galleguillos, D.; Forero, J.; Morales, L.C.; Lackey, S.W.; Kar, P.; di Pardo, A.; Holt, A.; Kerr, B.J.; Todd, K.G.; et al. Disease-modifying Effects of Ganglioside GM1 in Huntington’s Disease Models. EMBO Mol. Med. 2017, 9, 1537–1557. [Google Scholar] [CrossRef]
- Di Pardo, A.; Basit, A.; Armirotti, A.; Amico, E.; Castaldo, S.; Pepe, G.; Marracino, F.; Buttari, F.; Digilio, A.F.; Maglione, V. De Novo Synthesis of Sphingolipids Is Defective in Experimental Models of Huntington’s Disease. Front. Neurosci. 2017, 11, 698. [Google Scholar] [CrossRef]
- Yamout, B.I.; Alroughani, R. Multiple Sclerosis. Semin. Neurol. 2018, 38, 212–225. [Google Scholar] [CrossRef]
- Barthelmes, J.; de Bazo, A.M.; Pewzner-Jung, Y.; Schmitz, K.; Mayer, C.A.; Foerch, C.; Eberle, M.; Tafferner, N.; Ferreirós, N.; Henke, M.; et al. Lack of Ceramide Synthase 2 Suppresses the Development of Experimental Autoimmune Encephalomyelitis by Impairing the Migratory Capacity of Neutrophils. Brain Behav. Immun. 2015, 46, 280–292. [Google Scholar] [CrossRef]
- Eberle, M.; Ebel, P.; Mayer, C.A.; Barthelmes, J.; Tafferner, N.; Ferreiros, N.; Ulshöfer, T.; Henke, M.; Foerch, C.; de Bazo, A.M.; et al. Exacerbation of Experimental Autoimmune Encephalomyelitis in Ceramide Synthase 6 Knockout Mice Is Associated with Enhanced Activation/Migration of Neutrophils. Immunol. Cell Biol. 2015, 93, 825–836. [Google Scholar] [CrossRef]
- Schiffmann, S.; Ferreiros, N.; Birod, K.; Eberle, M.; Schreiber, Y.; Pfeilschifter, W.; Ziemann, U.; Pierre, S.; Scholich, K.; Grösch, S.; et al. Ceramide Synthase 6 Plays a Critical Role in the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2012, 188, 5723–5733. [Google Scholar] [CrossRef] [Green Version]
- Kurz, J.; Brunkhorst, R.; Foerch, C.; Blum, L.; Henke, M.; Gabriel, L.; Ulshöfer, T.; Ferreirós, N.; Parnham, M.J.; Geisslinger, G.; et al. The Relevance of Ceramides and Their Synthesizing Enzymes for Multiple Sclerosis. Clin. Sci. 2018, 132, 1963–1976. [Google Scholar] [CrossRef] [Green Version]
- Vidaurre, O.G.; Haines, J.D.; Sand, I.K.; Adula, K.P.; Huynh, J.L.; Mcgraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal Fluid Ceramides from Patients with Multiple Sclerosis Impair Neuronal Bioenergetics. Brain 2014, 137, 2271–2286. [Google Scholar] [CrossRef]
- Sashindranath, M.; Nandurkar, H.H. Endothelial Dysfunction in the Brain: Setting the Stage for Stroke and Other Cerebrovascular Complications of Covid-19. Stroke 2021, 52, 1895–1904. [Google Scholar] [CrossRef]
- Kahl, A.; Blanco, I.; Jackman, K.; Baskar, J.; Mohan, H.M.; Rodney-Sandy, R.; Zhang, S.; Iadecola, C.; Hochrainer, K. Cerebral Ischemia Induces the Aggregation of Proteins Linked to Neurodegenerative Diseases. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Kuźma, E.; Lourida, I.; Moore, S.F.; Levine, D.A.; Ukoumunne, O.C.; Llewellyn, D.J. Stroke and Dementia Risk: A Systematic Review and Meta-Analysis. Alzheimer’s Dement. 2018, 14, 1416–1426. [Google Scholar] [CrossRef] [Green Version]
- Dmitrieva, V.G.; Torshina, E.V.; Yuzhakov, V.V.; Povarova, O.V.; Skvortsova, V.I.; Limborska, S.A.; Dergunova, L.V. Expression of Sphingomyelin Synthase 1 Gene in Rat Brain Focal Ischemia. Brain Res. 2008, 1188, 222–227. [Google Scholar] [CrossRef]
- Yu, Z.F.; Nikolova-Karakashian, M.; Zhou, D.; Cheng, G.; Schuchman, E.H.; Mattson, M.P. Pivotal Role for Acidic Sphingomyelinase in Cerebral Ischemia-Induced Ceramide and Cytokine Production, and Neuronal Apoptosis. J. Mol. Neurosci. 2000, 15, 85–97. [Google Scholar] [CrossRef]
- Hagemann, N.; Yusuf, A.M.; Martiny, C.; Zhang, X.; Kleinschnitz, C.; Gunzer, M.; Kolesnick, R.; Gulbins, E.; Hermann, D.M. Homozygous Smpd1 Deficiency Aggravates Brain Ischemia/ Reperfusion Injury by Mechanisms Involving Polymorphonuclear Neutrophils, Whereas Heterozygous Smpd1 Deficiency Protects against Mild Focal Cerebral Ischemia. Basic Res. Cardiol. 2020, 115, 1–14. [Google Scholar] [CrossRef]
- Chao, H.C.; Lee, T.H.; Chiang, C.S.; Yang, S.Y.; Kuo, C.H.; Tang, S.C. Sphingolipidomics Investigation of the Temporal Dynamics after Ischemic Brain Injury. J. Proteome Res. 2019, 18, 3470–3478. [Google Scholar] [CrossRef]
- Gui, Y.-K.; Li, Q.; Liu, L.; Zeng, P.; Ren, R.F.; Guo, Z.F.; Wang, G.H.; Song, J.G.; Zhang, P. Plasma Levels of Ceramides Relate to Ischemic Stroke Risk and Clinical Severity. Brain Res. Bull. 2020, 158, 122–127. [Google Scholar] [CrossRef]
- Lee, T.H.; Cheng, C.N.; Chao, H.C.; Lee, C.H.; Kuo, C.H.; Tang, S.C.; Jeng, J.S. Plasma Ceramides Are Associated with Outcomes in Acute Ischemic Stroke Patients. J. Formos. Med. Assoc. 2021. [Google Scholar] [CrossRef]
- Tea, M.N.; Poonnoose, S.I.; Pitson, S.M. Targeting the Sphingolipid System as a Therapeutic Direction for Glioblastoma. Cancers 2020, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- Bernhart, E.; Damm, S.; Wintersperger, A.; Nusshold, C.; Brunner, A.M.; Plastira, I.; Rechberger, G.; Reicher, H.; Wadsack, C.; Zimmer, A.; et al. Interference with Distinct Steps of Sphingolipid Synthesis and Signaling Attenuates Proliferation of U87MG Glioma Cells. Biochem. Pharmacol. 2015, 96, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Casasampere, M.; Ordóñez, Y.F.; Casas, J.; Fabrias, G. Dihydroceramide Desaturase Inhibitors Induce Autophagy via Dihydroceramide-Dependent and Independent Mechanisms. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 264–275. [Google Scholar] [CrossRef]
- Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide Accumulation Mediates Cytotoxic Autophagy of Cancer Cells via Autolysosome Destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef] [Green Version]
- Van Brooklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine Kinase-1 Expression Correlates with Poor Survival of Patients with Glioblastoma Multiforme: Roles of Sphingosine Kinase Isoforms in Growth of Glioblastoma Cell Lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705. [Google Scholar] [CrossRef]
- Kapitonov, D.; Allegood, J.C.; Mitchell, C.; Hait, N.C.; Almenara, J.A.; Adams, J.K.; Zipkin, R.E.; Dent, P.; Kordula, T.; Milstien, S.; et al. Targeting Sphingosine Kinase 1 Inhibits Akt Signaling, Induces Apoptosis, and Suppresses Growth of Human Glioblastoma Cells and Xenografts. Cancer Res. 2009, 69, 6915–6923. [Google Scholar] [CrossRef] [Green Version]
- Greenamyre, J.T.; Betarbet, R.; Sherer, T.B. The Rotenone Model of Parkinson’s Disease: Genes, Environment and Mitochondria. Parkinsonism Relat. Disord. 2003, 9, 59–64. [Google Scholar] [CrossRef]
- Schneider, J.S. MPTP-induced Parkinsonism: Acceleration of Biochemical and Behavioral Recovery by GM1 Ganglioside Treatment. J. Neurosci. Res. 1992, 31, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L. Selective Loss of Glucocerebrosidase Activity in Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurodegener. 2015, 10, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P. Cerebrospinal Fluid β-Glucocerebrosidase Activity Is Reduced in Parkinson’s Disease Patients. Mov. Disord. 2009, 34, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase Activity in Parkinson’s Disease with and without GBA Mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced Glucocerebrosidase Is Associated with Increased α-Synuclein in Sporadic Parkinson’s Disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Jeon, S.; Burbulla, L.F.; Krainc, D. Acid Ceramidase Inhibition Ameliorates α-Synuclein Accumulation upon Loss of GBA1 Function. Hum. Mol. Genet. 2018, 27, 1972–1988. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E. Glucosylceramide Synthase Inhibition Alleviates Aberrations in Synucleinopathy Models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A Modulator of Wild-Type Glucocerebrosidase Improves Pathogenic Phenotypes in Dopaminergic Neuronal Models of Parkinson’s Disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther. 2017, 25, 2727–2742. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hayes, M.A.; Beagan, J.; McLean, J.R.; Izen, S.C.; Perez-Torres, E.; et al. Glucocerebrosidase Gene Therapy Prevents α-Synucleinopathy of Midbrain Dopamine Neurons. Neurobiol. Dis. 2015, 82, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA Deficiency Promotes SNCA/α-Synuclein Accumulation through Autophagic Inhibition by Inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, A.; Saddoughi, S.A.; Song, P.; Sultan, I.; Ponnusamy, S.; Senkal, C.E.; Snook, C.F.; Arnold, H.K.; Sears, R.C.; Hanniui, Y.A.; et al. Direct Interaction between the Inhibitor 2 and Ceramide via Sphingolipid-protein Binding Is Involved in the Regulation of Protein Phosphatase 2A Activity and Signaling. FASEB J. 2009, 23, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Salinas, M.; López-Valdaliso, R.; Martín, D.; Alvarez, A.; Cuadrado, A. Inhibition of PKB/Akt1 by C2-Ceramide Involves Activation of Ceramide- Activated Protein Phosphatase in PC12 Cells. Mol. Cell. Neurosci. 2000, 15, 156–169. [Google Scholar] [CrossRef]
- Cornell, T.T.; Hinkovska-Galcheva, V.; Sun, L.; Cai, Q.; Hershenson, M.B.; Vanway, S.; Shanley, T.P. Ceramide-Dependent PP2A Regulation of TNFα-Induced IL-8 Production in Respiratory Epithelial Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L849–L856. [Google Scholar] [CrossRef] [Green Version]
- Lleó, A.; Núñez-Llaves, R.; Alcolea, D.; Chiva, C.; Balateu-Paños, D.; Colom-Cadena, M.; Gomez-Giro, G.; Muñoz, L.; Querol-Vilaseca, M.; Pegueroles, J.; et al. Changes in Synaptic Proteins Precede Neurodegeneration Markers in Preclinical Alzheimer’s Disease Cerebrospinal Fluid*. Mol. Cell. Proteom. 2019, 18, 546–560. [Google Scholar] [CrossRef] [Green Version]
- Launey, T.; Endo, S.; Sakai, R.; Harano, J.; Ito, M. Protein Phosphatase 2A Inhibition Induces Cerebellar Long-Term Depression and Declustering of Synaptic AMPA Receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Hu, X.D.; Yang, H.; Xia, H. Distinct Roles of Protein Phosphatase 1 Bound on Neurabin and Spinophilin and Its Regulation in AMPA Receptor Trafficking and LTD Induction. Mol. Neurobiol. 2018, 55, 7179–7186. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Prieto, G.A.; Cotman, C.W. IL-1β Suppresses CLTP-Induced Surface Expression of GluA1 and Actin Polymerization via Ceramide-Mediated Src Activation. J. Neuroinflamm. 2018, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Mattson, M.P. The Transcription Factor NF-ΚB Mediates Increases in Calcium Currents and Decreases in NMDA- and AMPA/Kainate-Induced Currents Induced by Tumor Necrosis Factor-α in Hippocampal Neurons. J. Neurochem. 1998, 70, 1876–1886. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-Mediated Macroautophagy Involves Inhibition of Protein Kinase B and Up-Regulation of Beclin 1. J. Biol. Chem. 2004, 279, 18384–18391. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.P.T.; Tsika, E.; Kelly, K.; Levine, N.; Chen, X.; West, A.B.; Boularand, S.; Barneoud, P.; Moore, D.J. Dopaminergic Neurodegeneration Induced by Parkinson’s Disease-Linked G2019S LRRK2 Is Dependent on Kinase and GTPase Activity. Proc. Natl. Acad. Sci. USA 2020, 117, 17296–17307. [Google Scholar] [CrossRef]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and Mitochondria: Recent Advances and Current Views. Brain Res. 2019, 1702, 96–104. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson Disease: Challenges of Clinical Trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 Kinase Activity Alters Neuronal Autophagy by Disrupting the Axonal Transport of Autophagosomes. Curr. Biol. 2021, 31, 2140–2154.e6. [Google Scholar] [CrossRef]
- Yahalom, G.; Greenbaum, L.; Israeli-Korn, S.; Fay-Karmon, T.; Livneh, V.; Ruskey, J.A.; Roncière, L.; Alam, A.; Gan-Or, Z.; Hassin-Baer, S. Carriers of Both GBA and LRRK2 Mutations, Compared to Carriers of Either, in Parkinson’s Disease: Risk Estimates and Genotype-Phenotype Correlations. Parkinsonism Relat. Disord. 2019, 62, 179–184. [Google Scholar] [CrossRef]
- Ortega, R.A.; Wang, C.; Raymond, D.; Bryant, N.; Scherzer, C.R.; Thaler, A.; Alcalay, R.N.; West, A.B.; Mirelman, A.; Kuras, Y.; et al. Association of Dual LRRK2 G2019S and GBA Variations with Parkinson Disease Progression. JAMA Netw. Open 2021, 4, e215845. [Google Scholar] [CrossRef]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 Deficiency Impacts Ceramide Metabolism in Brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 Kinase Activity Regulates Lysosomal Glucocerebrosidase in Neurons Derived from Parkinson’s Disease Patients. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Weindel, C.G.; Bell, S.L.; Vail, K.J.; West, K.O.; Patrick, K.L.; Watson, R.O. LRRK2 Maintains Mitochondrial Homeostasis and Regulates Innate Immune Responses to Mycobacterium Tuberculosis. eLife 2020, 9, e56214. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Bonet-Ponce, L.; Blesa, J.R.; Romero, F.J.; Murphy, M.P.; Jordan, J.; Galindo, M.F. The LRRK2 Inhibitor GSK2578215A Induces Protective Autophagy in SH-SY5Y Cells: Involvement of Drp-1-Mediated Mitochondrial Fission and Mitochondrial-Derived ROS Signaling. Cell Death Dis. 2014, 5, e1368. [Google Scholar] [CrossRef] [Green Version]
- Fugio, L.B.; Coeli-Lacchini, F.B.; Leopoldino, A.M. Sphingolipids and Mitochondrial Dynamic. Cells 2020, 9, 581. [Google Scholar] [CrossRef] [Green Version]
- Van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.W.; Plasterk, R.H.A.; Nollen, E.A.A.C. Elegans Model Identifies Genetic Modifiers of α-Synuclein Inclusion Formation during Aging. PLoS Genet. 2008, 4, e1000212. [Google Scholar] [CrossRef]
- Khateeb, S.; Flusser, H.; Ofir, R.; Shelef, I.; Narkis, G.; Vardi, G.; Shorer, Z.; Levy, R.; Galil, A.; Elbedour, K.; et al. PLA2G6 Mutation Underlies Infantile Neuroaxonal Dystrophy. Am. J. Hum. Genet. 2006, 79, 942–948. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, H.; Yoshino, H.; Ogaki, K.; Li, L.; Yamashita, C.; Li, Y.; Funayama, M.; Sasaki, R.; Kokubo, Y.; Kuzuhara, S.; et al. PLA2G6 Variant in Parkinson’s Disease. J. Hum. Genet. 2011, 56, 401–403. [Google Scholar] [CrossRef]
- Shen, T.; Hu, J.; Jiang, Y.; Zhao, S.; Lin, C.; Yin, X.; Yan, Y.; Pu, J.; Lai, H.Y.; Zhang, B. Early-Onset Parkinson’s Disease Caused by Pla2g6 Compound Heterozygous Mutation, a Case Report and Literature Review. Front. Neurol. 2019, 10, 915. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N.; Kirber, M.T.; Mostoslavsky, G.; Ikezu, T.; Wolozin, B.; et al. Impairment of PARK14-Dependent Ca2+ Signalling Is a Novel Determinant of Parkinson’s Disease. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Lee, P.-T.; Chen, K.; Lin, W.-W.; Wang, L.; Bellen Correspondence, H.J. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to Alpha-Synuclein Gain. Cell Metab. 2018, 28, 605–618. [Google Scholar] [CrossRef] [Green Version]
- Follett, J.; Norwood, S.J.; Hamilton, N.A.; Mohan, M.; Kovtun, O.; Tay, S.; Zhe, Y.; Wood, S.A.; Mellick, G.D.; Silburn, P.A.; et al. The Vps35 D620N Mutation Linked to Parkinson’s Disease Disrupts the Cargo Sorting Function of Retromer. Traffic 2014, 15, 230–244. [Google Scholar] [CrossRef]
- Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceição, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carriço, J.A.; Alcalay, R.N. Serum Lipid Alterations in GBA-Associated Parkinson’s Disease. Parkinsonism Relat. Disord. 2017, 44, 58–65. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin Mitochondrial Quality Control: A Source of Regional Vulnerability in Parkinson’s Disease. Mol. Neurodegener. 2020, 15, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagishi, Y.; Nakano, N.; Ogino, M.; Ichimura, M.; Minami, A.; Matsuda, S. PINK1 Signaling in Mitochondrial Homeostasis and in Aging (Review). Int. J. Mol. Med. 2017, 39, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, M.; Nakashima, S.; Kiyono, T.; Nakagawa, M.; Yamada, J.; Yamakawa, H.; Banno, Y.; Shinoda, J.; Nishimura, Y.; Nozawa, Y.; et al. P53 Regulates Ceramide Formation by Neutral Sphingomyelinase through Reactive Oxygen Species in Human Glioma Cells. Oncogene 2001, 20, 1368–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Fernández-Checa, J.C. Direct Effect of Ceramide on the Mitochondrial Electron Transport Chain Leads to Generation of Reactive Oxygen Species: Role of Mitochondrial Glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef] [Green Version]
- Ausman, J.; Abbade, J.; Ermini, L.; Farrell, A.; Tagliaferro, A.; Post, M.; Caniggia, I. Ceramide-Induced BOK Promotes Mitochondrial Fission in Preeclampsia. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Qi, Z.; Yang, W.; Liu, Y.; Cui, T.; Gao, H.; Duan, C.; Lu, L.; Zhao, C.; Zhao, H.; Yang, H. Loss of PINK1 Function Decreases PP2A Activity and Promotes Autophagy in Dopaminergic Cells and a Murine Model. Neurochem. Int. 2011, 59, 572–581. [Google Scholar] [CrossRef]
- Dobrowsky, R.T.; Kamibayashi, C.; Mumby, M.C.; Hannuns, Y.A. Ceramide Activates Heterotrimeric Protein Phosphatase 2A. J. Biol. Chem. 1993, 268, 15523–15530. [Google Scholar] [CrossRef]
- Malini, E.; Zampieri, S.; Deganuto, M.; Romanello, M.; Sechi, A.; Bembi, B.; Dardis, A. Role of LIMP-2 in the Intracellular Trafficking of β-Glucosidase in Different Human Cellular Models. FASEB J. 2015, 29, 3839–3852. [Google Scholar] [CrossRef] [Green Version]
- Usenko, T.S.; Bezrukova, A.I.; Bogdanova, D.A.; Kopytova, A.E.; Senkevich, K.A.; Gracheva, E.V.; Timofeeva, A.A.; Miliukhina, I.V.; Zakharova, E.Y.; Emelyanov, A.; et al. Genetics Variants and Expression of the SCARB2 Gene in the Pathogenesis of Parkinson’s Disease in Russia. Neurosci. Lett. 2021, 741, 135509. [Google Scholar] [CrossRef]
- Bras, J.; Guerreiro, R.; Darwent, L.; Parkkinen, L.; Ansorge, O.; Escott-Price, V.; Hernandez, D.G.; Nalls, M.A.; Clark, L.N.; Honig, L.S.; et al. Genetic Analysis Implicates APOE, SNCA and Suggests Lysosomal Dysfunction in the Etiology of Dementia with Lewy Bodies. Hum. Mol. Genet. 2014, 23, 6139–6146. [Google Scholar] [CrossRef] [Green Version]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-Based Genome-Wide Association Study Identifies Two Novel Loci and a Substantial Genetic Component for Parkinson’s Disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; von Coelln, R.; Pihlstrøm, L.; Simón-Sánchez, J.; Schulte, C.; Sharma, M.; Krohn, L.; et al. Parkinson’s Disease Age at Onset Genome-Wide Association Study: Defining Heritability, Genetic Loci, and α-Synuclein Mechanisms. Mov. Disord. 2019, 34, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; Destefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Matrone, C.; Dzamko, N.; Madsen, P.; Nyegaard, M.; Pohlmann, R.; Søndergaard, R.V.; Lassen, L.B.; Andresen, T.L.; Halliday, G.M.; Jensen, P.H.; et al. Mannose 6-Phosphate Receptor Is Reduced in -Synuclein Overexpressing Models of Parkinsons Disease. PLoS ONE 2016, 11, e0160501. [Google Scholar] [CrossRef] [Green Version]
- Rothaug, M.; Zunke, F.; Mazzulli, J.R.; Schweizer, M.; Altmeppen, H.; Lüllmann-Rauche, R.; Kallemeijn, W.W.; Gaspar, P.; Aerts, J.M.; Glatzel, M.; et al. LIMP-2 Expression Is Critical for β-Glucocerebrosidase Activity and α-Synuclein Clearance. Proc. Natl. Acad. Sci. USA 2014, 111, 15573–15578. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.L.; Zhang, C.W.; Li, L. The Sources of Reactive Oxygen Species and Its Possible Role in the Pathogenesis of Parkinson’s Disease. Parkinson’s Dis. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Radad, K.; Al-Shraim, M.; Al-Emam, A.; Wang, F.; Kranner, B.; Rausch, W.D.; Moldzio, R. Rotenone: From Modelling to Implication in Parkinson’s Disease. Folia Neuropathol. 2019, 57, 317–326. [Google Scholar] [CrossRef]
- Magrassi, L.; Adorni, L.; Montorfano, G.; Rapelli, S.; Butti, G.; Berra, B.; Milanesi, G. Vitamin D Metabolites Activate the Sphingomyelin Pathway and Induce Death of Glioblastoma Cells. Acta Neurochir. 1998, 140, 707–714. [Google Scholar] [CrossRef]
- Pyszko, J.A.; Strosznajder, J.B. The Key Role of Sphingosine Kinases in the Molecular Mechanism of Neuronal Cell Survival and Death in an Experimental Model of Parkinson’s Disease. Folia Neuropathol. 2014, 52, 260–269. [Google Scholar] [CrossRef]
- Pyszko, J.; Strosznajder, J.B. Sphingosine Kinase 1 and Sphingosine-1-Phosphate in Oxidative Stress Evoked by 1-Methyl-4-Phenylpyridinium (MPP+) in Human Dopaminergic Neuronal Cells. Mol. Neurobiol. 2014, 50, 38–48. [Google Scholar] [CrossRef]
- Shao, Q.-H.; Chen, Y.; Li, F.-F.; Wang, S.; Zhang, X.-L.; Yuan, Y.-H.; Chen, N.-H. TLR4 Deficiency Has a Protective Effect in the MPTP/Probenecid Mouse Model of Parkinson’s Disease. Acta Pharmacol. Sin. 2019, 40, 1503–1512. [Google Scholar] [CrossRef]
- Zhao, X.D.; Wang, F.X.; Cao, W.F.; Zhang, Y.H.; Li, Y. TLR4 Signaling Mediates AP-1 Activation in an MPTP-Induced Mouse Model of Parkinson’s Disease. Int. Immunopharmacol. 2016, 32, 96–102. [Google Scholar] [CrossRef]
- Mariucci, G.; Pagiotti, R.; Galli, F.; Romani, L.; Conte, C. The Potential Role of Toll-Like Receptor 4 in Mediating Dopaminergic Cell Loss and Alpha-Synuclein Expression in the Acute MPTP Mouse Model of Parkinson’s Disease. J. Mol. Neurosci. 2018, 64, 611–618. [Google Scholar] [CrossRef]
- Conte, C.; Roscini, L.; Sardella, R.; Mariucci, G.; Scorzoni, S.; Beccari, T.; Corte, L. Toll Like Receptor 4 Affects the Cerebral Biochemical Changes Induced by MPTP Treatment. Neurochem. Res. 2017, 42, 493–500. [Google Scholar] [CrossRef]
- Kaur, G.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Kumar, A.; Arora, S.; Bungau, S. Role of TLR2 and TLR4 Signaling in Parkinson’s Disease: An Insight into Associated Therapeutic Potential. J. Mol. Neurosci. MN 2021, 1–16. [Google Scholar] [CrossRef]
- Albi, E.; Cataldi, S.; Codini, M.; Mariucci, G.; Lazzarini, A.; Ceccarini, M.R.; Ferri, I.; Laurenti, M.E.; Arcuri, C.; Patria, F.; et al. Neutral Sphingomyelinase Increases and Delocalizes in the Absence of Toll-Like Receptor 4: A New Insight for MPTP Neurotoxicity. Prostaglandins Other Lipid Mediat. 2019, 142, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Chen, M.; Du, T.; Duan, C.; Gao, G.; Yang, H. The Novel Mechanism of Rotenone-Induced α-Synuclein Phosphorylation via Reduced Protein Phosphatase 2A Activity. Int. J. Biochem. Cell Biol. 2016, 75, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Induction of Apoptotic Erythrocyte Death by Rotenone. Toxicology 2012, 300, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Rutkute, K.; Asmis, R.H.; Nikolova-Karakashian, M.N. Regulation of Neutral Sphingomyelinase-2 by GSH: A New Insight to the Role of Oxidative Stress in Aging-Associated Inflammation. J. Lipid Res. 2007, 48, 2443–2452. [Google Scholar] [CrossRef] [Green Version]
- Merrill, A.H. Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef]
- Grammatikos, G.; Schoell, N.; Ferreirós, N.; Bon, D.; Herrmann, E.; Farnik, H.; Köberle, V.; Piiper, A.; Zeuzem, S.; Kronenberger, B.; et al. Serum Sphingolipidomic Analyses Reveal an Upregulation of C16-Ceramide and Sphingosine-1-Phosphate in Hepatocellular Carcinoma. Oncotarget 2016, 7, 18095–18105. [Google Scholar] [CrossRef] [Green Version]
- Alberg, A.J.; Armeson, K.; Pierce, J.S.; Bielawski, J.; Bielawska, A.; Visvanathan, K.; Hill, E.G.; Ogretmen, B. Plasma Sphingolipids and Lung Cancer: A Population-Based, Nested Case-Control Study. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1374–1382. [Google Scholar] [CrossRef] [Green Version]
- Govindarajah, N.; Clifford, R.; Bowden, D.; Sutton, P.A.; Parsons, J.L.; Vimalachandran, D. Sphingolipids and Acid Ceramidase as Therapeutic Targets in Cancer Therapy. Crit. Rev. Oncol. Hematol. 2019, 138, 104–111. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Bonte, M.A.; Desplanque, M.; Gressier, B.; Devos, D.; Chartier-Harlin, M.C. Glycosphingolipids and Neuroinflammation in Parkinson’s Disease. Mol. Neurodegener. 2020, 15, 1–16. [Google Scholar] [CrossRef]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered Ceramide Acyl Chain Length and Ceramide Synthase Gene Expression in Parkinson’s Disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Irigoyen, J.; Cartas-Cejudo, P.; Iruarrizaga-Lejarreta, M.; Santamaría, E. Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study. Biomedicines 2021, 9, 491. [Google Scholar] [CrossRef]
- Xing, Y.; Tang, Y.; Zhao, L.; Wang, Q.; Qin, W.; Ji, X.; Zhang, J.; Jia, J. Associations between Plasma Ceramides and Cognitive and Neuropsychiatric Manifestations in Parkinson’s Disease Dementia. J. Neurol. Sci. 2016, 370, 82–87. [Google Scholar] [CrossRef]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.-K.; Ber-Sultan, S.G.; Schleicher, E.; et al. Plasma Ceramide and Glucosylceramide Metabolism Is Altered in Sporadic Parkinson’s Disease and Associated with Cognitive Impairment: A Pilot Study. PLoS ONE 2013, 9, e0073094. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Jenner, A.M.; Shui, G.; Cheong, W.F.; Mitchell, T.W.; Nealon, J.R.; Kim, W.S.; McCann, H.; Wenk, M.R.; Halliday, G.M. Lipid Pathway Alterations in Parkinson’s Disease Primary Visual Cortex. PLoS ONE 2011, 6, e0017299. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H.V. No Evidence for Substrate Accumulation in Parkinson Brains with GBA Mutations. Mov. Disord. 2015, 30, 1085–1089. [Google Scholar] [CrossRef]
- Stoessel, D.; Schulte, C.; Santos, M.C.; Scheller, D.; Rebollo-Mesa, I.; Deuschle, C.; Walther, D.; Schauer, N.; Berg, D.; Costa, A. Promising Metabolite Profiles in the Plasma and CSF of Early Clinical Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 1–14. [Google Scholar] [CrossRef]
- Boutin, M.; Sun, Y.; Shacka, J.J.; Auray-Blais, C. Tandem Mass Spectrometry Multiplex Analysis of Glucosylceramide and Galactosylceramide Isoforms in Brain Tissues at Different Stages of Parkinson Disease. Anal. Chem. 2016, 88, 1856–1863. [Google Scholar] [CrossRef]
- Wood, P.L.; Tippireddy, S.; Feriante, J.; Woltjer, R.L. Augmented Frontal Cortex Diacylglycerol Levels in Parkinson’s Disease and Lewy Body Disease. PLoS ONE 2018, 13, e0191815. [Google Scholar] [CrossRef]
- Niimi, Y.; Mizutani, Y.; Akiyama, H.; Watanabe, H.; Shiroki, R.; Hirabayashi, Y.; Hoshinaga, K.; Mutoh, T. Cerebrospinal Fluid Profiles in Parkinson’s Disease: No Accumulation of Glucosylceramide, but Significant Downregulation of Active Complement C5 Fragment. J. Parkinson’s Dis. 2021, 11, 221–232. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice Lacking Major Brain Gangliosides Develop Parkinsonism. Neurochem. Res. 2011, 36, 1706–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.C.; Choo, Y.K.; Jou, I.; Kim, J.; Park, S.M. On the Mechanism of Internalization of α-Synuclein into Microglia: Roles of Ganglioside GM1 and Lipid Raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 Ganglioside Modifies α-Synuclein Toxicity and Is Neuroprotective in a Rat α-Synuclein Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, R.; Pallbo, J.; Weininger, U.; Linse, S.; Sparr, E. Ganglioside Lipids Accelerate α-Synuclein Amyloid Formation. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 1062–1072. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of Ganglioside GM1 Correlates with Parkinson’s Disease in Mice and Humans. J. Neurosci. Res. 2012, 90, 1997–2008. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Wang, L.; Yang, C. High Performance Liquid Chromatography-Mass Spectrometry (LC-MS) Based Quantitative Lipidomics Study of Ganglioside-NANA-3 Plasma to Establish Its Association with Parkinson’s Disease Patients. Med. Sci. Monit. 2017, 23, 5345–5353. [Google Scholar] [CrossRef]
- Chan, R.B.; Perotte, A.J.; Zhou, B.; Liong, C.; Shorr, E.J.; Marder, K.S.; Kang, U.J.; Waters, C.H.; Levy, O.A.; Xu, Y.; et al. Elevated GM3 Plasma Concentration in Idiopathic Parkinson’s Disease: A Lipidomic Analysis. PLoS ONE 2017, 12, e0172348. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Custodia, A.; Aramburu-Núñez, M.; Correa-Paz, C.; Posado-Fernández, A.; Gómez-Larrauri, A.; Castillo, J.; Gómez-Muñoz, A.; Sobrino, T.; Ouro, A. Ceramide Metabolism and Parkinson’s Disease—Therapeutic Targets. Biomolecules 2021, 11, 945. https://doi.org/10.3390/biom11070945
Custodia A, Aramburu-Núñez M, Correa-Paz C, Posado-Fernández A, Gómez-Larrauri A, Castillo J, Gómez-Muñoz A, Sobrino T, Ouro A. Ceramide Metabolism and Parkinson’s Disease—Therapeutic Targets. Biomolecules. 2021; 11(7):945. https://doi.org/10.3390/biom11070945
Chicago/Turabian StyleCustodia, Antía, Marta Aramburu-Núñez, Clara Correa-Paz, Adrián Posado-Fernández, Ana Gómez-Larrauri, José Castillo, Antonio Gómez-Muñoz, Tomás Sobrino, and Alberto Ouro. 2021. "Ceramide Metabolism and Parkinson’s Disease—Therapeutic Targets" Biomolecules 11, no. 7: 945. https://doi.org/10.3390/biom11070945
APA StyleCustodia, A., Aramburu-Núñez, M., Correa-Paz, C., Posado-Fernández, A., Gómez-Larrauri, A., Castillo, J., Gómez-Muñoz, A., Sobrino, T., & Ouro, A. (2021). Ceramide Metabolism and Parkinson’s Disease—Therapeutic Targets. Biomolecules, 11(7), 945. https://doi.org/10.3390/biom11070945