1. Introduction
Helicases are proteins that unwind nucleic acids and remodel protein-nucleic acid complexes in a wide spectrum of cellular tasks. DNA helicases are critical in maintaining cellular integrity by playing important roles in DNA replication, recombination and repair. RNA helicases are likewise fundamental by orchestrating transcription, RNA processing, ribosome biogenesis, translation and RNA turnover. Helicases are classified into 6 superfamilies (SF1-6) [
1]. The SF1-6 share a common helicase core with a set of helicase signature motifs. The SF2 is the largest and most diverse group of helicases with more than ten families. SF2 members are non-hexameric helicases that share a conserved helicase core with nine characteristic motifs and that often contain N- and/or C-terminal accessory domains involved in the regulation of their activities [
2,
3]. The core provides the active site for ATP hydrolysis, binds nucleic acid and performs a basal unwinding activity. Although ATP-dependent unwinding of nucleic acid duplexes is their hallmark reaction, not all helicases catalyse unwinding in vitro, and disrupt duplexes in vivo [
4,
5]. Among SF2 helicases, the Lhr (Large helicase related) proteins are scarcely characterized. They are found in some Bacteria but are ubiquitous in Archaea [
4,
5]. To date, no homologs of Lhr proteins have been reported in Eukarya.
In Bacteria, Lhr proteins are mostly prevalent in Proteobacteria and Actinobacteria. Lhr proteins from
Pseudomonas putida (
Pput),
Escherichia coli (
Ecol) and
Mycobacterium smegmatis (
Msme) are among the few helicases that have been characterized [
6,
7,
8,
9]. The 1507 amino acid (aa)
Msme-Lhr is the founding member of the Lhr helicase family [
9]. The crystal structure of
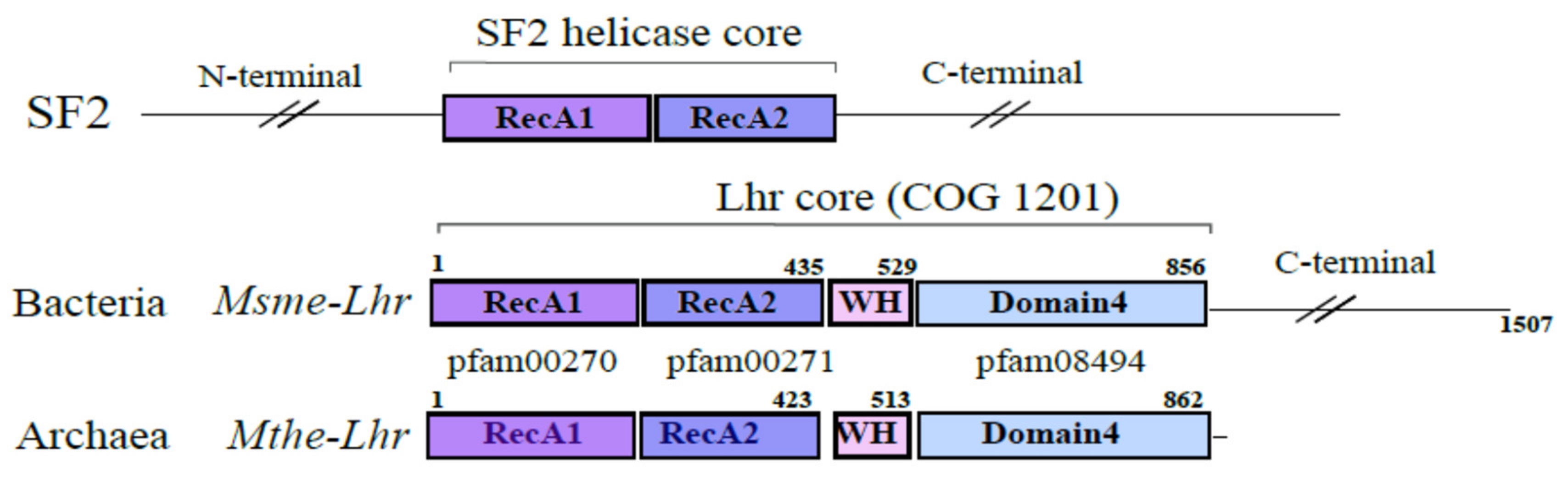
Msme-Lhr restricted to the first 856 aa was solved and uncovered a specific structural domain organization also referred to as the “Lhr-Core”: two RecA domains in tandem (RecA1 and RecA2), a winged-helix (WH) motif and a domain annotated as Domain 4 whose function is still unknown. Interestingly, the WH displays a similar fold to the one observed in Hjm and RecQ (also called Hel308) DNA helicases [
9,
10]. While
Pput-Lhr is restricted to the “Lhr-Core”,
Msme-Lhr and
Ecol-Lhr have an additional C-terminal domain [
6,
7,
8,
9] (
Figure 1).
The studies characterizing the biochemical activities and the functions of bacterial Lhr proteins have mainly revealed a role of Lhr helicases in DNA repair. Nonetheless, some of their properties suggest that Lhr may also participate in RNA processing. In vivo, the gene encoding
Msme-Lhr was shown to be upregulated when cells were exposed to DNA damaging agents [
11,
12]. Regarding
Ecol-Lhr, though its deletion does not increase cell-sensitivity to UV or H
2O
2 [
8], Cooper et al. demonstrated a synthetic genetic interaction with RadA, a RecA-related protein involved in the processing of recombination intermediates [
13]. In vitro,
Pput-Lhr and
Msme-Lhr helicases were shown to have DNA-dependent ATPase and ATP-dependent 3′-to-5′ translocase activities. While
Pput-Lhr exhibits no preference for DNA:DNA or DNA:RNA duplex [
7],
Msme-Lhr prefers to unwind DNA:RNA duplexes in which the displaced strand is RNA [
6]. Finally, the importance of
Eco
l-Lhr rose from its occurrence in a cluster with the gene encoding RNase T, a ribonuclease involved in the maturation of stable RNAs, as well as in DNA repair pathways [
8]. This interaction occurs at the transcriptional level, as the Lhr and RNase T are co-transcribed, but no interaction at the protein level was reported yet either in vitro or in vivo.
In Archaea, some genome annotations record two types of Lhr proteins, called here aLhr1 and aLhr2. The aLhr1 and aLhr2 exhibit a “Lhr-Core” domain organization [
14]. aLhr1 has an additional cysteine-rich motif at its C-terminal end. Only a few Lhr from Sulfolobales (TACK) and Methanobacteriales (Euryarchaea) have been studied [
15,
16]. Lhr of
Sulfolobus islandicus (SiRe_1605) was found to be important for the transcription of genes in nucleotide metabolism and DNA repair [
17]. Monomeric Lhr of
Sulfolobus solfataricus, also known as Hel112, was characterized in vitro as an ATP-dependent DNA helicase with a 3′-5′ polarity and a preference for forked DNA substrates [
16]. Lhr of
Sulfolobus acidocaldarius (saci_1500, also named RecQ-like helicase) was found to be important for DNA repair after UV-induced stress [
18]. In vitro, Lhr of
Methanothermobacter thermautotrophicus (
Mthe) was also found to have 3′-5′ directional DNA translocase activity and to act on forked DNA structures. In a genetic assay, its expression gave a phenotype identical to the DNA helicases Hel308 and RecQ involved in replication-coupled DNA repair [
15]. In addition, aLhr2 of
Pyrococcus abyssi (
Paby) was detected in the interaction network of proteins implicated in DNA replication and repair [
19]. Recently, we also spotted
Paby-aLhr2 as a partner of players in RNA metabolism. Indeed,
Paby-aLhr2 was identified in the interaction network of the RNA helicase ASH-Ski2 together with the 5′-3′ and 3′-5′ RNA degradation machineries, aRNase J and the RNA exosome, respectively [
20] (
Table S1). It should be noted that DNA metabolism enzymes were also identified in this network. This raises questions about the role of the aLhr2 proteins in Thermococcales.
In this study, we highlighted archaeal Lhr-type proteins as ubiquitous enzymes by revisiting the Lhr-type proteins landscape, using in-depth phylogenomic analyses. We identified six distinct phylogenetic groups of Lhr proteins, three in Archaea and three in Bacteria. We also defined the phylogenetic groups to which each of the experimentally studied Lhr helicases belong to. To go further in understanding the relevance of the archaeal aLhr2 group members in DNA and/or RNA metabolism, we characterized the enzymatic properties of aLhr2 from the Thermococcales Thermococcus barophilus (Tbar-aLhr2). Our results allowed us to propose that Tbar-aLhr2 is a DNA/RNA helicase with significant annealing activity that acts on DNA:RNA hybrids and on RNA:RNA duplexes.
2. Materials & Methods
2.1. Building Lhr-Type Dataset
Completely sequenced genomes of 286 Archaea and 3769 Bacteria showing a high level of annotation were downloaded from EBI (
http://www.ebi.ac.uk/genomes/; accessed on 3 May 2019). The complete genomes of these 4055 strains, their proteomes and EMBL features were managed with an in-house MySQL database. Moreover, we had previously performed the annotation of the protein sequences of these genomes against the conserved domain database downloaded from the NCBI (
https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml; accessed on 2 April 2019) using the
rpsblast program [
21]. The
hmmscan program [
22] was used to annotate proteins with Pfam (32.0) domains. To avoid redundancies due to multiple repetitions of strains of the same species, we retained only one strain per species. Conversely, in order to obtain a better coverage of Archaea’s diversity, 75 Asgard proteomes were retrieved from UniProt, although they do not have the same sequencing and annotation quality as the other archaeal genomes (35 Lokiarchaeota, 27 Thorarchaeota, 12 Heimdallarchaeota and one Odinarchaeota). An initial sample of 1381 Lhr protein candidates were identified using the COG1201 (Lhr-like helicase) annotation performed by
rpsblast. In order to eliminate the false positives while keeping the most divergent sequences, we set an e-value threshold ≤ 1 × e
−04 associated with an alignment covering of at least 30% of the COG.
To identify Lhr-like families, we performed all-against-all
blastp comparisons of our initial set of proteins with default parameters, except for
-max_target_seqs which was set to 1381 sequences. The results were filtered to retain only the best bi-directional hits between proteins of different species. Protein relationships were then converted into a graph in which the vertices represent protein sequences, and the edges represent their relationships [
23]. The edges were weighted by the average pairwise -log
10 E-value. The graph was further processed by a graph-partitioning approach based on the Markov Clustering algorithm (MCL, [
24]). The inflate factor (IF) value is an important parameter of MCL as it regulates the cluster granularity. We tested several IF values (from 2 to 6) and a partitioning into 9 stable classes was observed starting from an IF ≥ 4. Classes 1 to 9 have sizes of 615, 344, 220, 106, 68, 22, 3, 1, and 1 sequences, respectively. A single protein from
Pseudomonas viridiflava (
A0A1Y6JKR4_PSEVI) was not classified and was discarded as a false positive since it shared only a small region of similarity with PF00271.
In order to facilitate phylogenetic reconstructions while preserving the diversity of sequences in the original sample, we represented sequences with more than 70% identity by a single sequence, the medoid. To achieve this, the edges of the previous graph with an identity < 70% were removed. This pruned graph was further processed by MCL to identify groups of closely related sequences (identity ≥ 70). This identified 352 groups (including 27 Asgard groups) composed of a unique sequence and 111 groups composed of many closely related sequences. For each of the 111 groups, we computed the medoid, i.e., the sequence with the minimal average dissimilarity to all the other proteins in the group. We added the constraint that its length should be close to the median length of all sequences of the group. This resulted in a set of 463 proteins composed of 352 unique sequences and 111 medoids. Eight unique sequences were discarded as they did not have the PF00270 (DEAD) and/or the PF00271 (Helicase_C) domains. The aLhr1 and aLhr2 sequences of
T. barophilus belong to two groups of closely related sequences. As a result of our sample size reduction process, these two sequences were not selected as medoid. The selected medoids were aLhr1 from
Thermococcus profundus (TproA01.ASJ02541.1) and aLhr2 from
Methanocaldococcus jannaschii (MjanA01. AAB98279.1). As aLhr2 from
T. barophilus is the subject of this experimental study, the
Tbar-aLhr1 (TbarA01.ADT83607.1) and
Tbar-aLhr2 (TbarA01.ADT83510.1) protein sequences were added back to our sample. The tree in
Figure 2 (Results section) shows that each sequence of
T. barophilus has a direct common ancestor with their respective medoid. Our final sample contains 457 sequences (
Supplementary Table S2A,B).
Since the Sfth helicases appear to be the closest related family of Lhr helicases, both families sharing a common ancestor [
14], we used Sfth sequences to root our Lhr family tree. To build the Sfth sample, we applied a similar strategy as the one described above for Lhr protein identification. Sfth protein candidates were identified using the COG1205 annotation performed by
rpsblast. Proteins that did not possess the three expected domains (PF00270 DEAD; PF09369 DUF1998; PF00271 Helicase_C) were excluded. Using an identity threshold of 55%, we obtained 24 medoid sequences that we further used as representatives of this family (
Supplementary Table S2C).
2.2. Alignment of the Core Helicase Domain
In order to eliminate the variability of the N- and C-term regions of the proteins, we extracted the central domain of the SF2 helicase core, i.e., the RecA1 and RecA2 regions (
Figure 1). The coordinates of the alignment of the sequences with the PF00270 and PF00271 domains were used to extract both regions, which were then merged for each sequence. These sequences were aligned with
mafft [
25] (parameters: -reorder -localpair -maxiterate 1000). In order to improve the quality of the alignments and to keep as much information as possible, we used the
divvier method [
26] with the option
-divvy (full divvying) and
-mincol 4. This strategy was applied to the dataset containing only Lhr sequences and to the dataset composed of Lhr and Sfth sequences.
2.3. Protein Family and Archaeal Species Trees
The best-fit amino acid substitution model for the data was selected with
modeltest-ng [
27] and the phylogenetic trees were inferred using the
iq-tree software [
28]. The same best model was selected for both datasets (-m LG4M + I). Branch supports were measured with ultra-fast bootstrap approximations (
-bb 1000) and single branch tests (
-alrt 1000). The trees were annotated and visualized with the online tool Interactive Tree Of Life (iTOLv5,
https://itol.embl.de; accessed on 3 May 2019) [
29].
To construct the archaeal species tree, we used the 122 markers that have been identified as reliable for phylogenetic inference [
30]. We first thought of including the Asgard species in the tree but as their genomes are mostly partial, too many markers were missing for this to be feasible. Therefore, the tree was built by taking into account 219 archaeal species. The set of 122 protein markers was characterized by HMM profiles from the Pfam (v27) and the TIGRFAMs (v15.0) databases. For each genome included in this study, the proteins were identified by using each Pfam entry as query in the
hmmsearch program from the HMMER 3.3.1 package (downloaded from
http://hmmer.org/; accessed on 3 May 2019 [
22]) with the
--cut_tc (trusted cutoff) parameter. The
hmmsearch output domain file was parsed to extract, for each genome and for each HMM profile, the best protein hit. Protein alignments with HMM profiles were merged for each marker. We thus obtained 122 sequence alignments. The columns of the alignments that had a high deletion frequency were removed with
trimal (
-gt 0.1) [
31]. The quality of the alignments was estimated using the
t-coffee transitive consistency score (TCS) [
32]. The analysis of the results obtained on each alignment allowed us to (i) eliminate sequences with outlier TCS values and (ii) discard two alignments (PF04104.9 and PF01990.12) with a low overall TCS value (TCS < 65). The resulting 120 marker alignments were concatenated, the tree was inferred with
fasttree [
33] under the LG + GAMMA model, and branch support values were determined using 100 non-parametric bootstrap replicates. The tree was rooted on DPANN Archaea according to [
34].
2.4. Genomic Context Analysis
We extracted the proteins encoded by the genes located less than 4000 bp upstream and downstream from the predicted alhr2 genes. To obtain a functional characterization and classification of these proteins, they were annotated by hmmscan with TIGR HMM profiles. iTOL was used to associate these gene neighbourhoods to the species tree (DATASET_DOMAINS option).
2.5. Expression Vectors
The
Supplementary Table S3 summarizes the oligonucleotides used in this study. All constructions were obtained by assembling PCR fragments using InFusion
® cloning kit (Takara). Using an appropriate set of oligonucleotides, the pET11b (untagged protein) vector was linearized by PCR amplification with the PrimeSTAR Max DNA polymerase (Takara), and the coding sequences of
T. barophilus aLhr2 (TERMP_00533) and
P. abyssi Hel308 (PAB_0592) were amplified from genomic DNA with the Phusion High-Fidelity DNA polymerase (ThermoFisherScientific). The pET11b vectors expressing the aLhr2-T215A, aLhr2-W577A and aLhr2-I512A variants were generated by site-directed mutagenesis of their wild-type counterpart with appropriate sets of oligonucleotides using the QuikChange II XL Kit (Stratagene). The pET11b vectors expressing the truncated aLhr2-ΔDom4 and the Domain 4 by itself (aLhr2-Dom4) were constructed by reverse PCR on the pET11b-aLhr2-WT using specific phosphorylated oligonucleotides and by DNA ligation (T4 DNA ligase).
2.6. Purification of Tbar-aLhr2 Recombinant Proteins
E. coli BL21-CodonPlus (DE3) cells freshly transformed with pET11b-aLhr2, pET11b-aLhr2-T215A, pET11b-aLhr2-W577A, pET11b-aLhr2-I512A, pET11b-aLhr2-ΔDom4 and pET11b-aLhr2-Dom4 vectors were grown in 400 mL of LB medium at 37 °C. Protein production was induced at OD600 nm 0.8 with 0.2 mM IPTG. After 3 h of induction at 30 °C, the cells were collected, suspended in 10 mL of lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 10% glycerol) supplemented with 1 mg·mL−1 of lysozyme and a mix of EDTA-free protease inhibitor (cOmpleteTM, Roche, Merck KGaA, Darmstadt, Germany), and lysed by sonication (4 × [5 × 10 s], 50% cycle, VibraCell Biolock Scientific). The cleared extracts, obtained by centrifuging the crude extracts (20,000× g, 4 °C, 20 min), were treated with a mix of RNase A (20 µg·mL−1), RNase T1 (1 U·µL−1) and DNase I (20 µg·mL−1) containing 10 mM of MgCl2 for 30 min at 37 °C. After a heating step at 70 °C for 20 min, the extracts were further clarified by centrifugation (20,000× g, 4 °C, 20 min). First, the recombinant proteins were purified from the soluble fractions to near homogeneity using FPLC (Fast Protein Liquid Chromatography, Äkta-purifier10, GE-Healthcare) and specific columns (GE Healthcare): for wild type aLhr2, the punctual mutants, and aLhr2-Dom4, by a cation exchange chromatography (Hitrap SP HP); for aLhr2-ΔDom4 by a heparin column (Heparin FF) with a linear gradient of NaCl (300 mM to 1 M). Then, all recombinant proteins were loaded on a size-exclusion HiLoad 16/60 Superdex 200 PG column in 20 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol buffer.
2.7. Preparation of Radiolabelled Nucleic Acid Substrate
The 26-nt RNA (RNA
26) and all the DNA (DNA
26, DNA
31, DNA
50 and DNA
59) oligonucleotides were synthesized by Eurofins. The 50-nt RNA substrate (RNA
50) was obtained by in vitro transcription from a PCR fragment where DNA
50 was fused to the T7 promoter using the MEGAscript kit (Ambion). The DNA and RNA substrates were 5′-end radiolabelled using T4 polynucleotide kinase and γ-
32P-ATP. To prepare nucleic acid duplexes, the short DNA or RNA oligonucleotide was radiolabelled, mixed with an unlabelled DNA or RNA complementary strand at a 1:1 molar ratio (100 nM each), incubated for 5 min at 95 °C in 1X SSC buffer, and then slowly cooled at room temperature. The nucleotide sequences of all the substrates used in this study are given in
Supplementary Table S4.
2.8. ATPase Hydrolysis Assay
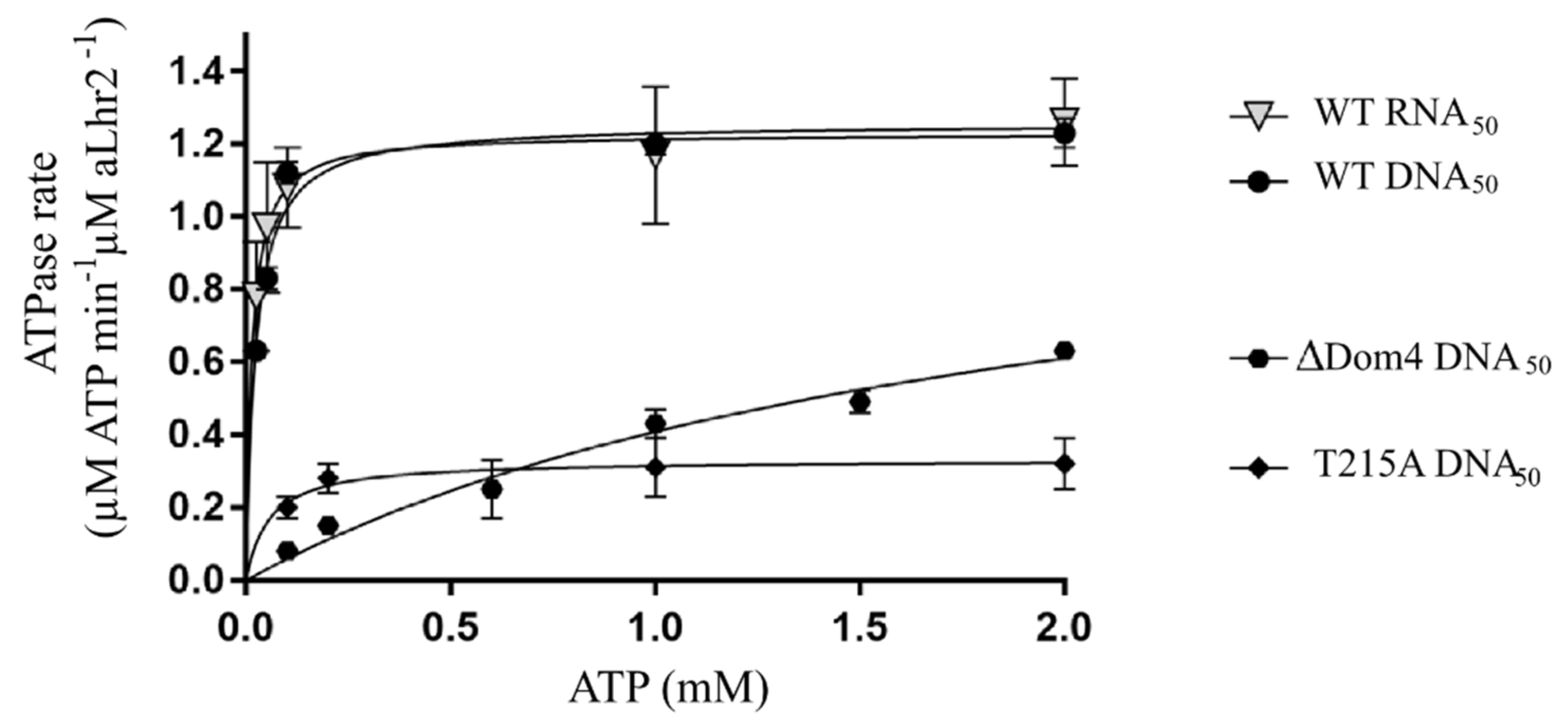
500 nM of recombinant protein were mixed with 5 nM of DNA50 or DNA59:DNA31 substrates in a 50 mM Hepes pH 7.5, 50 mM KCl, 5 mM MgCl2, 2 mM DTT buffer and preincubated for 10 min at 65 °C. 2 mM ATP and 0.85 µCi γ-32P-ATP were added at the 0 time point. The kinetic process was performed at 65 °C. At the indicated time, aliquots were spotted directly onto the TLC plate (PEI-cellulose, Macherey Nagel SAS, Hoerdt, France). TLC were developed with 0.25 M KH2PO4. Radioactive signals were measured using a PhosphorImager device (Typhoon Trio) and quantified with MultiGauge software (FujiFilm). The percentage of ATP versus ADP was plotted over time. Identical experiments were performed with 5 nM of DNA50 or RNA50 with a range of ATP concentration (0.025, 0.05, 0.1, 1, and 2 mM) in triplicates. The plots were derived using GraphPad Prism 7 software.
2.9. Nucleic Acid Binding Assay
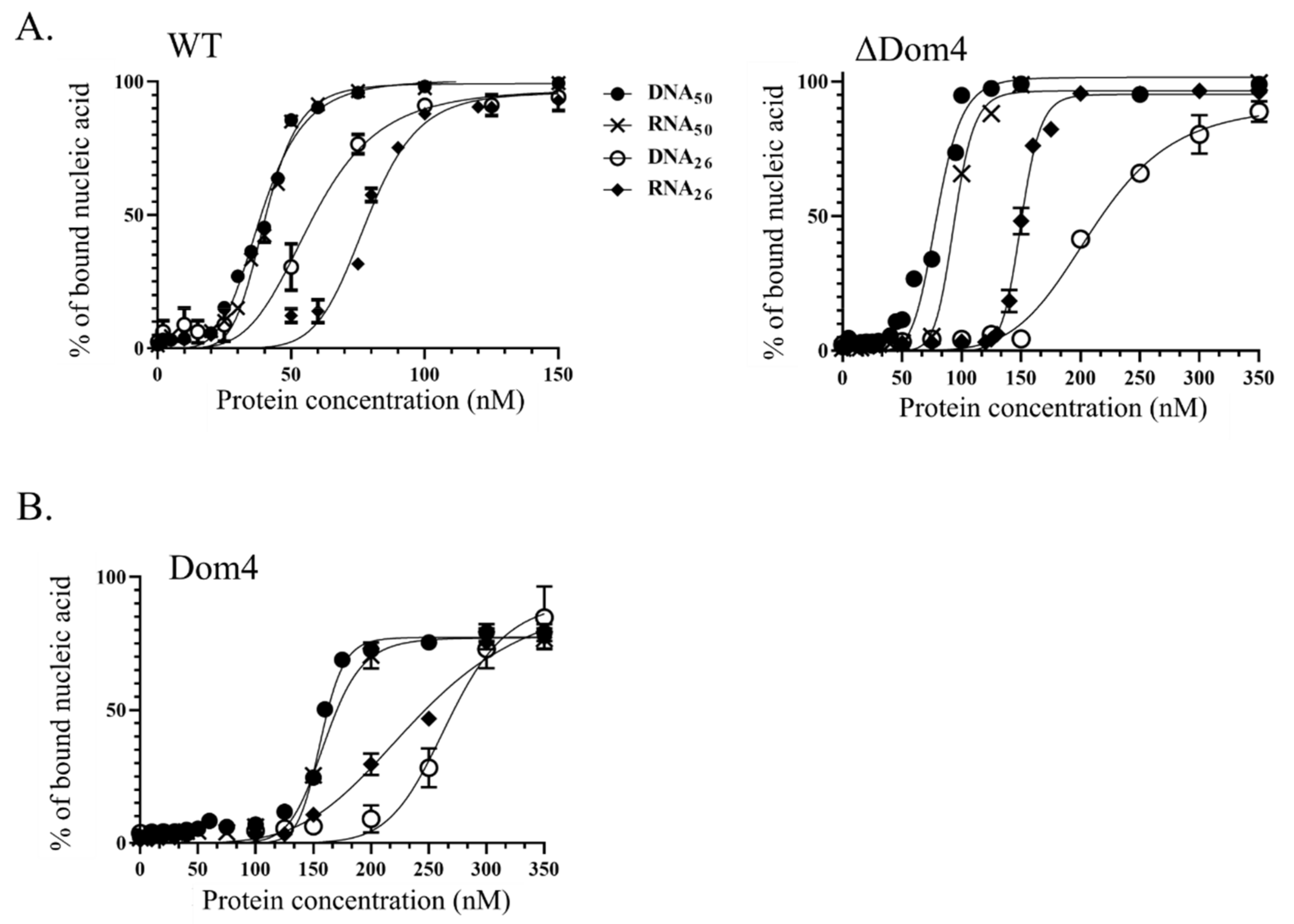
Double filtration binding assays were performed with range of protein concentrations from 0 to 350 nM and 0.5 nM of 32P-labelled RNA or DNA substrate using a Slot blot device (Amersham Biosciences). The protein was preincubated for 10 min at 65 °C in 25 mM Tris-HCl pH 8, 50 mM NaAc, 5 mM MgCl2, 2.5 mM β-Mercaptoethanol. After adding the substrate, the reactions were incubated for 15 min at 30 °C. Free nucleic acids were separated from nucleoprotein complexes on double filtration systems using Nylon and Nitrocellulose membranes (AmershamTM Hydond-N and Protran, respectively). Radioactive signals were measured using a PhosphorImager device and quantified with MultiGauge software. The apparent dissociation constants KD were calculated using GraphPad Prism 7 software.
The oligomerization state of
Tbar-aLhr2 was determined by size exclusion chromatography. After cation exchange chromatography (see
Section 2.6), the protein was concentrated and desalted using a Vivaspin centrifugal concentrator with a molecular weight cut-off of 50,000 Da (Sartorius). About 2 µM of protein was preincubated at 65 °C for 10 min in 25 mM Tris-HCl pH 7.5, 50 mM NaAc, 5 mM MgCl2, 300 mM NaCl. After adding 1 µM of DNA
50 substrate, the reaction was incubated for 15 min at 30 °C, and the mixture was loaded on a size-exclusion Superdex 200 Increase 10/300 GL column in 25 mM Tris-HCl pH 7.5, 50 mM NaAc, 5 mM MgCl2, 300 mM NaCl, 10% glycerol buffer. The fractions were analysed by Coomassie-blue SDS-PAGE and Western blotting.
2.10. Helicase (Unwinding) Assay
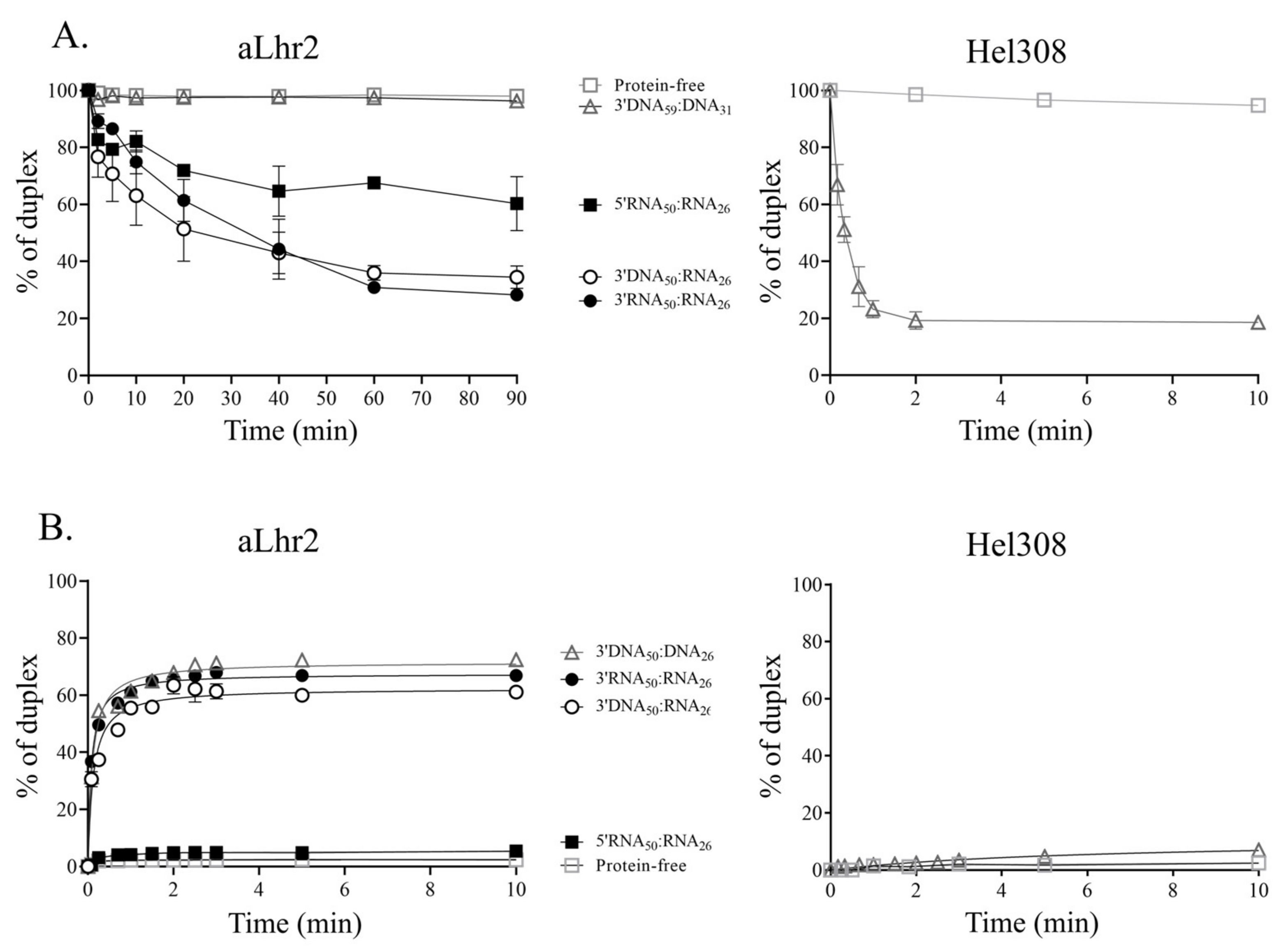
The unwinding assays were done with 250 nM of protein, 5 nM of α-32P-labeled nucleic acid duplex and a 200-fold excess of the unlabelled oligo trap (1 µM). The protein was preincubated separately for 5 min at 65 °C in 25 mM Tris-HCl pH 8, 50 mM NaAc, 2.5 mM β-Mercaptoethanol, 5 mM MgCl2, 25 mM ATP. After addition of the recombinant protein (250 nM), the reaction mixtures were incubated at 65 °C for the indicated times and then quenched with 0.5% SDS, 40 mM EDTA, 0.5 mg·mL−1 Proteinase K, 0.1% Bromophenol blue, and 20% glycerol. The reaction products were separated on a native 8% polyacrylamide gel (1X TBE, 0.1% SDS) by electrophoresis in 1X TBE (200Volts, 90 min). Radioactive signals were measured using a PhosphorImager device and quantified with MultiGauge software. All assays were repeated at least three times.
2.11. Strand-Annealing Assay
5 nM of radiolabelled substrates and 250 nM of recombinant protein were preincubated separately for 5 min at 65 °C in 25 mM Tris-HCl pH 8, 50 mM NaAc and 2.5 mM β-Mercaptoethanol. The reactions were started by mixing the protein and nucleic acid samples. After incubation at 65 °C, samples of 5 µL were withdrawn at the indicated time points. The reactions were quenched and analysed as described in
Section 2.10. All assays were independently repeated at least three times.
4. Discussion
Helicases are key enzymes involved in processes that depend on DNA and RNA transactions. They are known to use the energy of ATP to unwind nucleic acids and remodel protein-nucleic acid complexes. Here, we focused on the Thermococcales SF2 helicase aLhr2.
P. abyssi aLhr2 (
Paby-aLhr2) was found in the network of proteins involved in DNA replication and repair [
19]. However, our recent observation also found that
Paby-aLhr2 was part of the interaction network of proteins involved in RNA processing [
20]. This questions the function(s) of Thermococcales aLhr2. Since
Paby-aLhr2 is toxic when expressed in
E. coli, we investigated the in vitro activity of its orthologue in
T. barophilus (
Tbar-aLhr2). We determined that it is a monomeric DNA/RNA helicase able to process DNA:RNA and RNA:RNA duplexes. Moreover, for other archaeal aLhr helicases that were proposed to be DNA helicases involved in DNA repair and recombination [
15,
41], it was unclear if they belonged to the same aLhr2 group. We also interrogated the relationship that exists between the archaeal and bacterial Lhr proteins. Thus, we performed extensive phylogenomic analyses to elucidate the evolutionary links between Lhr proteins in Archaea and in Bacteria.
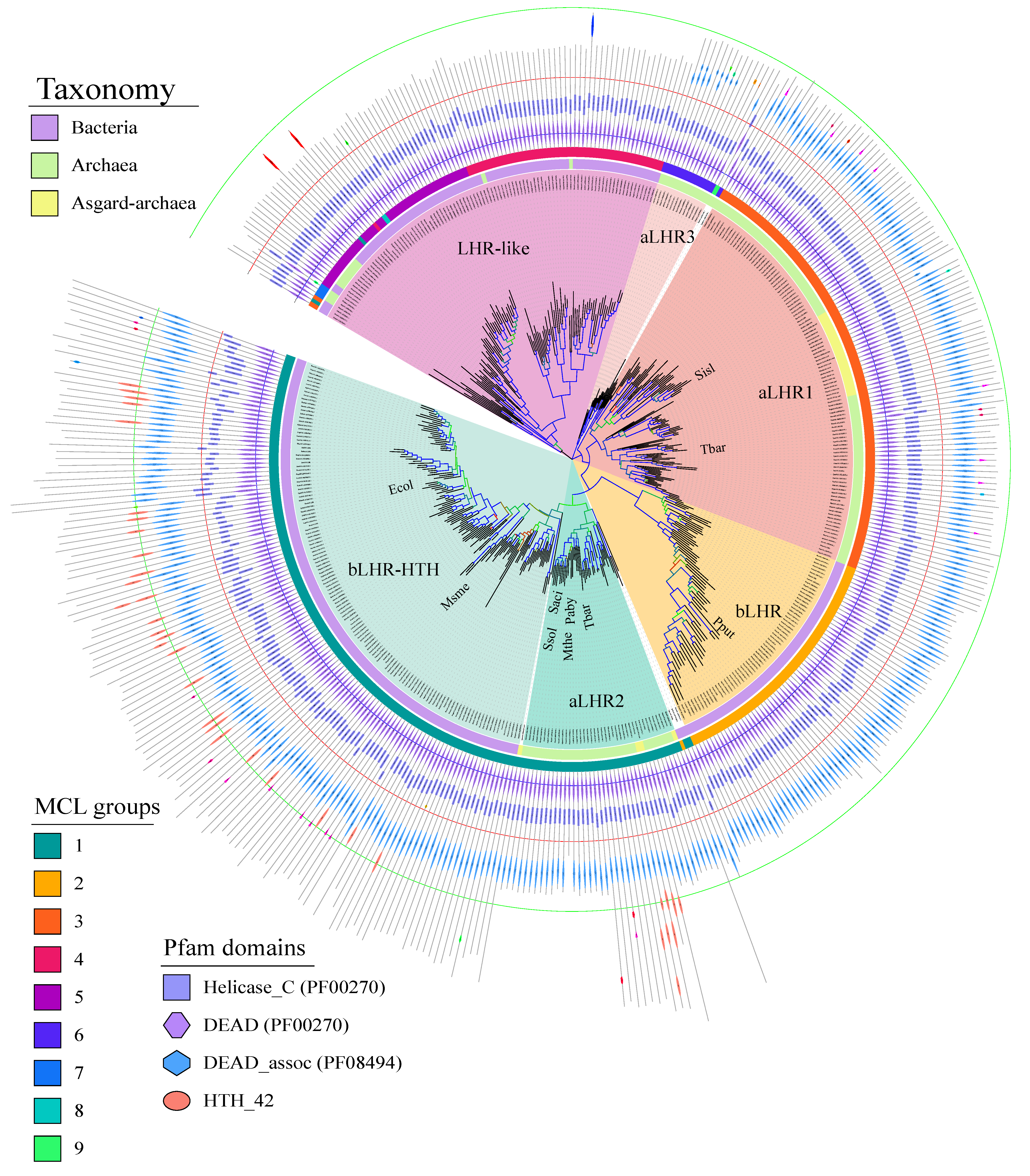
The Lhr-type proteins are defined by their unique domain organization [
9]. The “Lhr core” is composed of a SF2 helicase core, a winged-helix motif and an Lhr-specific Domain 4 (
Figure 1). After recovering and annotating the archaeal and bacterial Lhr-type proteins based on domain organization, we established their partition in MCL groups and computed a protein family tree with the conserved SF2 core region (
Figure 2). We could distinguish six groups sharing a common origin: three groups include only Lhr proteins from Archaea: aLhr1, aLhr2 and aLhr3; two groups include only Lhr proteins from Bacteria: bLhr and bLhr-HTH; and the last group, while dominated by Bacteria, includes few archaeal proteins (Lhr-like). The few archaeal sequences belonging to the Lhr-like group are scattered on the trees and should have been acquired from Bacteria through horizontal gene transfers. The Lhr groups were named based on previous studies [
14] and domain organization. According to the tree topology (
Figure 2), the sequences of the bLhr-HTH and aLhr2 groups share a common ancestor that predates the divergence of Bacteria and Archaea, suggesting that these sequences are orthologous and may have conserved similar roles in these genomes. The sequences of the bLhr group are characterized by significantly longer branches than those of the other groups. This reflects an acceleration in their rate of evolution and could be responsible for the instability of the anchoring of this group between the Sfth rooted (
Supplementary Figure S1) and Lhr-like rooted (
Figure 2) trees. Despite this difference in localization, the bLhr sequences share, on both trees, a common hypothetical ancestor with the bLhr-HTH and aLhr2 helicases. On the other hand, the aLhr1 group does not appear directly related to bacterial sequences.
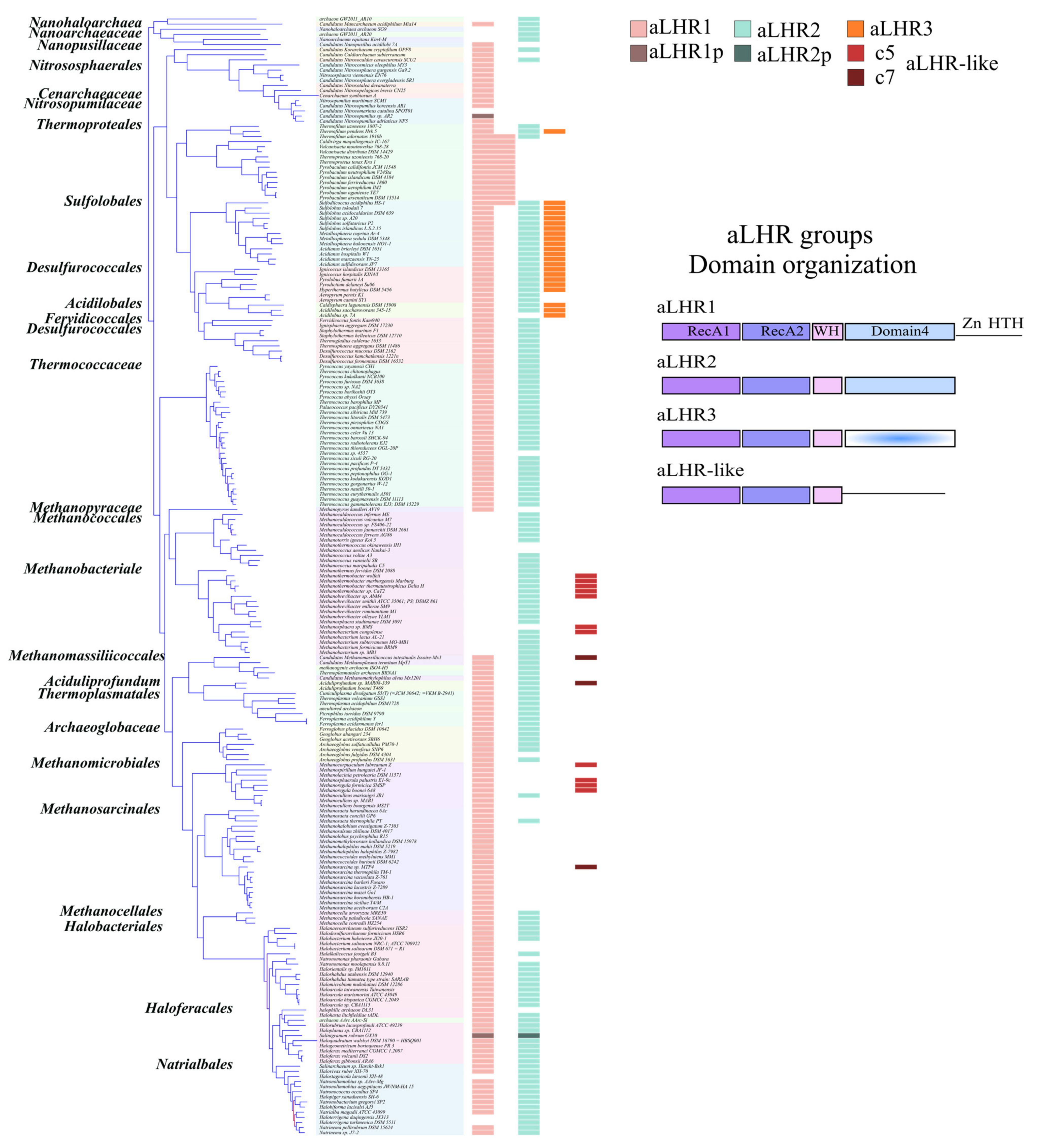
While initial work identified two groups of archaeal Lhr proteins, aLhr1 and aLhr2 [
14], we identified a third group aLhr3 that has a highly deteriorated Domain 4 and that seems to be limited to the Sulfolobales, Desulfurococcales and Acidobales. On the other hand, aLhr1 and aLhr2 are widespread in archaeal genomes and often present together. Their absence in some genomes would most likely result from different independent events of gene loss. Interestingly, only four out of 219 have lost both genes. We defined the characterized Lhr proteins of
S. solfataricus,
S. acidocaldarius, and
M. thermautotrophicus [
15,
16] to belong to the aLhr2 group. To date, no aLhr1 have been characterized. Genetic studies on a strain of
S. islandicus deleted the gene encoding aLhr1 [
17] suggesting that
Sisl-aLhr1 has a role in DNA repair, as shown for
Ssol-,
Saci- and
Mthe-aLhr2, but its biochemical properties are unknown. More work is needed to identify the common and/or specific functions of aLhr1 and aLhr2 in archaeal cells. In particular, since aLhr1 differs from aLhr2 by an additional cysteine-rich motif at its C-terminal end, the presence or not of an additional putative Zinc-finger might differentiate the proteins’ activities and partners.
Interestingly, in eleven genomes the RecA2 (10/11) or RecA1 (1/11) domain of aLhr2 is spliced by an intein_splicing domain. Proteins containing this integration are dispersed on the tree suggesting independent acquisitions. Interestingly, ATPase domains were shown to be hot-spots for inteins integration, with 70% of all inteins residing in ATPase-containing proteins, and at many different integration sites [
42]. While they are generally considered as being selfish parasites, intein splicing has recently been shown to be regulated by external stimuli such as temperature, pH, salt and DNA damage [
42]. Thus, some aLhr2 proteins might be regulated at the post-translational level and activated upon stress.
In Bacteria, for the first time, we identified three groups of Lhr proteins: bLhr, bLhr-HTH and Lhr-like. While bLhr proteins are restricted to the “Lhr core”, the group of bLhr-HTH has an additional HTH_42 domain at its C-terminal end. As before, the presence of an extra domain interrogates its role in the protein’s activities and interaction with partners. To date, one bLhr from
P. putida and two bLhr-HTH from
M. smegmatis and
E. coli have been studied, but the role of the HTH_42 domain has not been investigated. Interestingly, some aLhr2 from Methanomassilicoccales also possess an additional HTH_42 extension. Finally, the Lhr-like were defined as Lhr-type proteins, but while they possess a C-terminal region, no Domain 4 could be defined. Nonetheless, prediction of the structure of Lhr-likes from
Streptomyces coelicolor showed that ttheir C-terminal domain adopts a structure that is similar to the structure of Domain 4 of
Msme-bLhr-HTH (
Supplementary Figure S9).
In this study, we also report the in vitro activities of aLhr2 from
T. barophilus. First, we showed that the ATPase activity of
Tbar-aLhr2 is consistent with that measured for the bacterial
Msme-bLhr-HTH and
Pput-bLhr proteins [
6,
43]. We showed that
Tbar-aLhr2 is a nucleic acid-dependent ATPase with no apparent preference for DNA or RNA molecules (
Figure 5). Only the archaeal
Ssol-aLhr2 was shown to be able to hydrolyse ATP in the absence of nucleic acids [
41].
Ssol-aLhr2 also differs from the monomeric
Tbar-aLhr2 and bacterial Lhr proteins by its low affinity for single-stranded DNA and by its oligomeric state that can be both monomeric and dimeric; monomers and dimers having specific biochemical activities.
We also characterized
Tbar-aLhr2 as a monomeric DNA/RNA helicase able to process DNA:RNA and RNA:RNA duplexes (
Figure S4 &
Figure 7, left panels). Interestingly, we highlighted that the in vitro unwinding and annealing activities of
Tbar-aLhr2 differ drastically from those of Hel308, described as a DNA helicase involved in DNA repair [
35,
44]. Indeed, we showed that while
Tbar-aLhr2 is more prone to anneal nucleic strands than to unwind them,
Paby-Hel308 unwinds 3′DNA:DNA homoduplexes but does not form them from ssDNA molecules (
Figure 7). Altogether, these results clearly indicate that while both proteins are proposed as being involved in DNA repairs [
15,
40], they most likely perform different tasks in DNA transactions.
Among Lhr proteins, the capacity to process DNA:RNA hybrids is not specific to
Tbar-aLhr2. Indeed, while the bacterial
Msme-bLhr-HTH and
Pput-bLhr, and the archaeal
Mthe-aLhr2 were shown to process other substrates (forked DNA or Holliday junctions), they are also able to unwind DNA:RNA hybrids [
6,
7,
9,
15].
Msme-bLhr-HTH even prefers 3′-tailed RNA:DNA hybrids over DNA:DNA duplexes and was described as an RNA/DNA helicase [
9]. The capacity of
Tbar-aLhr2 to unwind more efficiently in vitro hybrids with a 3′ overhang strand that is indicative of a 3′ to 5′ polarity is also consistent with the polarity previously observed for its archaeal and bacterial counterparts [
6,
7,
9,
15,
16].
Tbar-aLhr2 has a significant ability to anneal single-stranded nucleic acid substrates to form DNA:DNA, RNA:RNA or RNA:DNA duplexes with no apparent preferences (
Figure 7). Both the monomeric and dimeric
Ssol-aLhr2 were also shown to have DNA strand annealing activities that are comparable to that of
Tbar-aLhr2 [
16]. Intriguingly, we found that
Tbar-aLhr2 is less prone to unwind duplexes (
Figure 7A) than to anneal nucleic strands (
Figure 7B). Indeed, we noted that the in vitro unwinding activity of
Tbar-aLhr2 is slow. This is also the case for
Msme-bLhr-HTH which was shown to have a capacity to only slowly dissociate nucleic acid strands (observed after 30 min of incubation) [
6]. This can be relevant for the cellular functions of
Tbar-aLhr2 but it can also mean that our experimental conditions are not optimal.
ATP binding is proposed to act as a molecular switch from a strand-annealing to an unwinding mode by changing the protein conformation. Here, we showed that
Tbar-aLhr2 unwinding activity is independent of the presence of ATP. While it might seem surprising, ATP-independent unwinding activities were previously reported for the human SF2 NS3h helicase and the bacterial SF1 RecBCD helicase [
45,
46]. It was proposed that the energy required for duplex separation is provided by nucleic acid binding and not by ATP binding and hydrolysis. In vivo, it is possible that the balance between unwinding and annealing states is displaced upon interaction with specific protein partners.
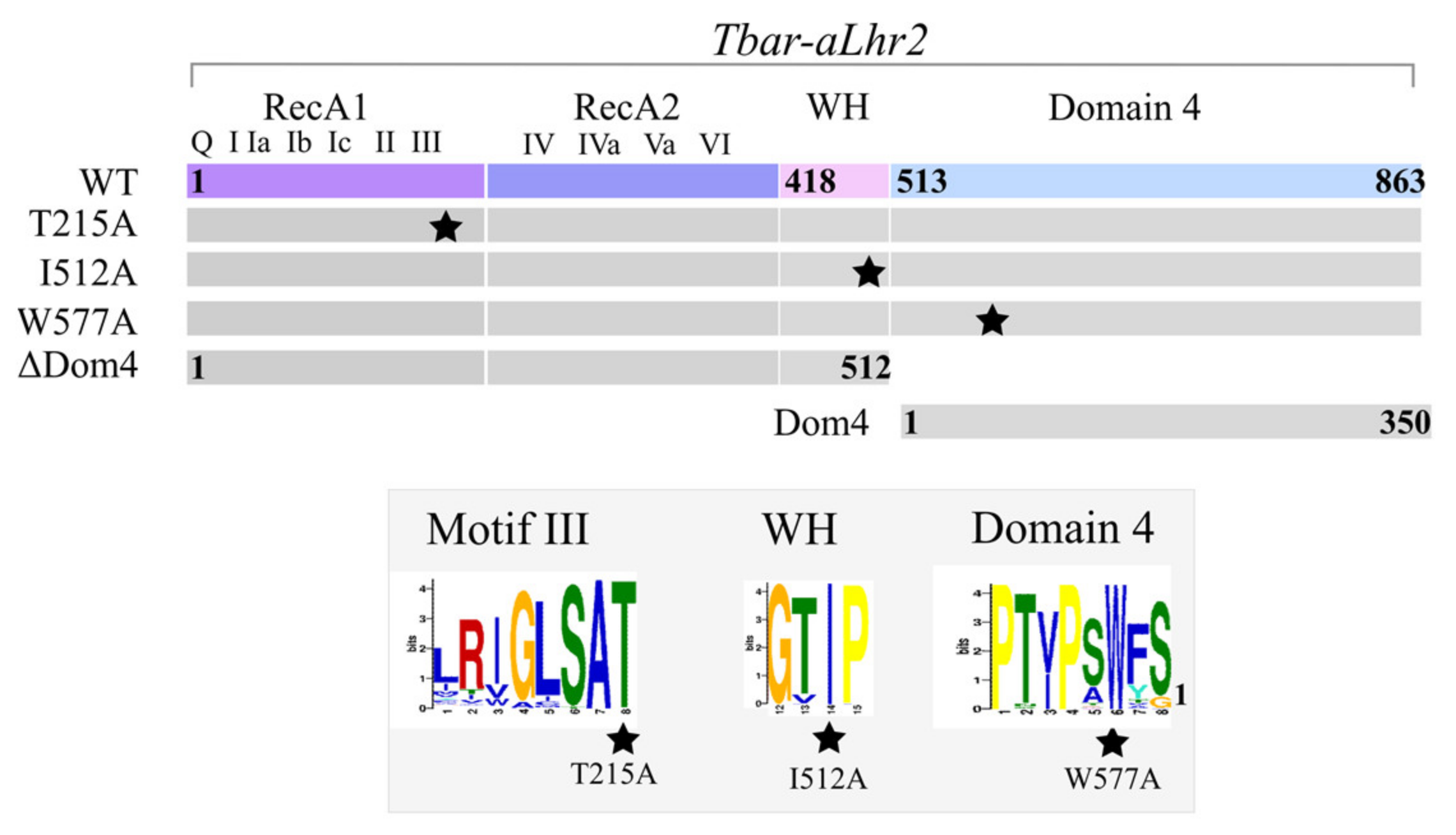
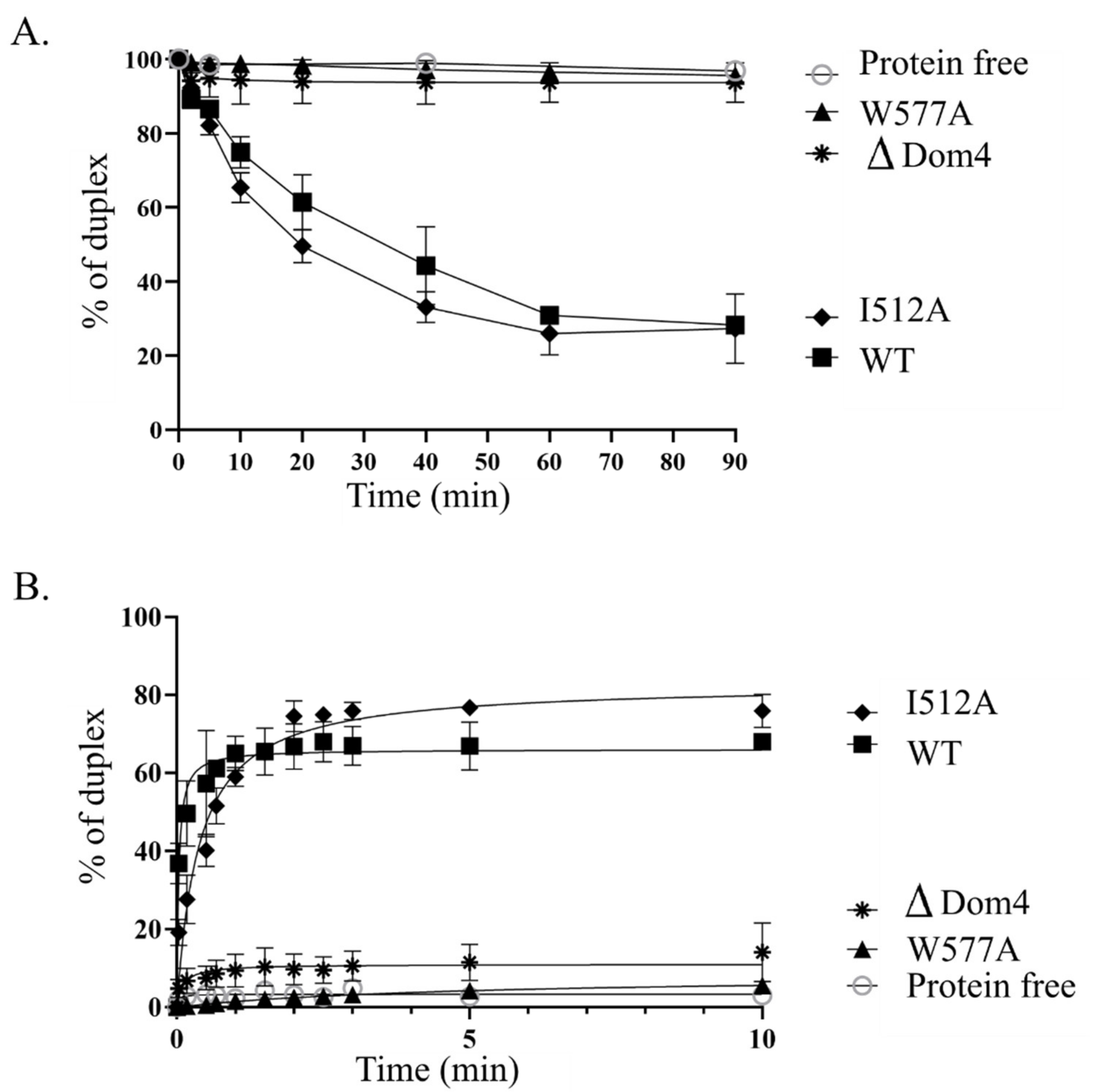
We also investigated the role of Domain 4, a novel structural domain specific to Lhr proteins, and demonstrated that it is essential for
Tbar-aLhr2 to adopt active conformations. First, we showed that while the ATPase activity is carried by the SF2 catalytic core composed of the RecA1 and RecA2 domains, Domain 4 stimulates ATP hydrolysis (
Supplementary Figure S4). Finally, we found that Domain 4 is essential for
Tbar-aLhr2 annealing and unwinding activities. The substitution of the highly conserved tryptophan at position 577 in
Tbar-aLhr2 Domain 4 is sufficient to abolish these reactions (
Figure 8). These results are in agreement with those obtained for bacterial
Msme-bLhr-HTH restricted to the “Lhr core” [
9].
Our results underline the high capacity of
Tbar-aLhr2 to perform nucleic acid strand annealing. This property could be of great importance in hyperthermophile organisms, such as
T. barophilus, for maintaining nucleic acid duplexes at high temperatures. While there is much that remains to be discovered with respect to the cellular functions of aLhr2 in vivo, we can propose the following roles for Thermococcales aLhr2. First, the detection of
Paby-aLhr2 in the interaction network of the replication protein A complex (RPA) [
19] is consistent with studies proposing that aLhr2 helicases are involved in DNA recombination and repair in
S. solfataricus and
M. thermautotrophicus [
15,
41]. Indeed, RPA that binds ssDNA is crucial for both DNA replication and DNA damage response [
47]. This is also coherent with an involvement of Thermococcales aLhr2 in RNA transactions. In
P. abyssi, RPA was also shown to enhance transcription [
19] and to be part of the interaction network of 5′-3′ exoribonuclease aRNase J [
20], questioning its involvement in RNA metabolism. The involvement of Thermococcales aLhr2 in RNA metabolism is also supported by our initial observation that
Paby-aLhr2 was found to be in the protein network of ASH-Ski2 with a high specificity index (
Supplementary Table S1); ASH-Ski2 is an archaeal specific Ski2-like helicase that forms a complex with aRNase J [
20]. Interestingly, RPA is also found in the interaction network of ASH-Ski2 (
Supplementary Table S1).
Furthermore, we showed that
Tbar-aLhr2 is a DNA/RNA helicase able to process DNA:RNA duplexes. The ability of Lhr proteins to process such hybrids was also identified for bacterial
Msme-bLhr-HTH and
Pput-bLhr, and for archaeal
Mthe-aLhr2 helicases [
6,
7,
9,
15]. In the cells, DNA:RNA hybrids are often found in the R-loop, a three-stranded structure that harbours a DNA:RNA hybrid and a displaced single-stranded DNA. Controlling R-loop formation and suppression is critical for many cellular processes. While R-loops are often associated with genome instability, DNA damage and transcription elongation defects, mounting evidence suggest that R-loops promote DNA transactions including DNA recombination and repair [
48]. Interestingly, RPA was also recently revealed to act as a sensor of R-loops and to regulate RNase H1 in human cells [
49]. Moreover, defect in mRNA processing was recently associated with R-loop-dependent genome instability in Eukaryotes [
48]. Further physiological and mechanical studies are necessary to determine the function(s) of aLhr2 in Thermococcales cells.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}