
Angiotensin II-Induced Vasoconstriction via Rho Kinase Activation in Pressure-Overloaded Rat Thoracic Aortas


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Abdominal Aortic Banding Operation
2.3. Organ Chamber Experiments
2.4. Western Blotting
2.5. Statistical Analysis
3. Results
3.1. Ang II-Induced Contraction in the Thoracic Aorta of Sham-Operated and Pressure-Overloaded Rats
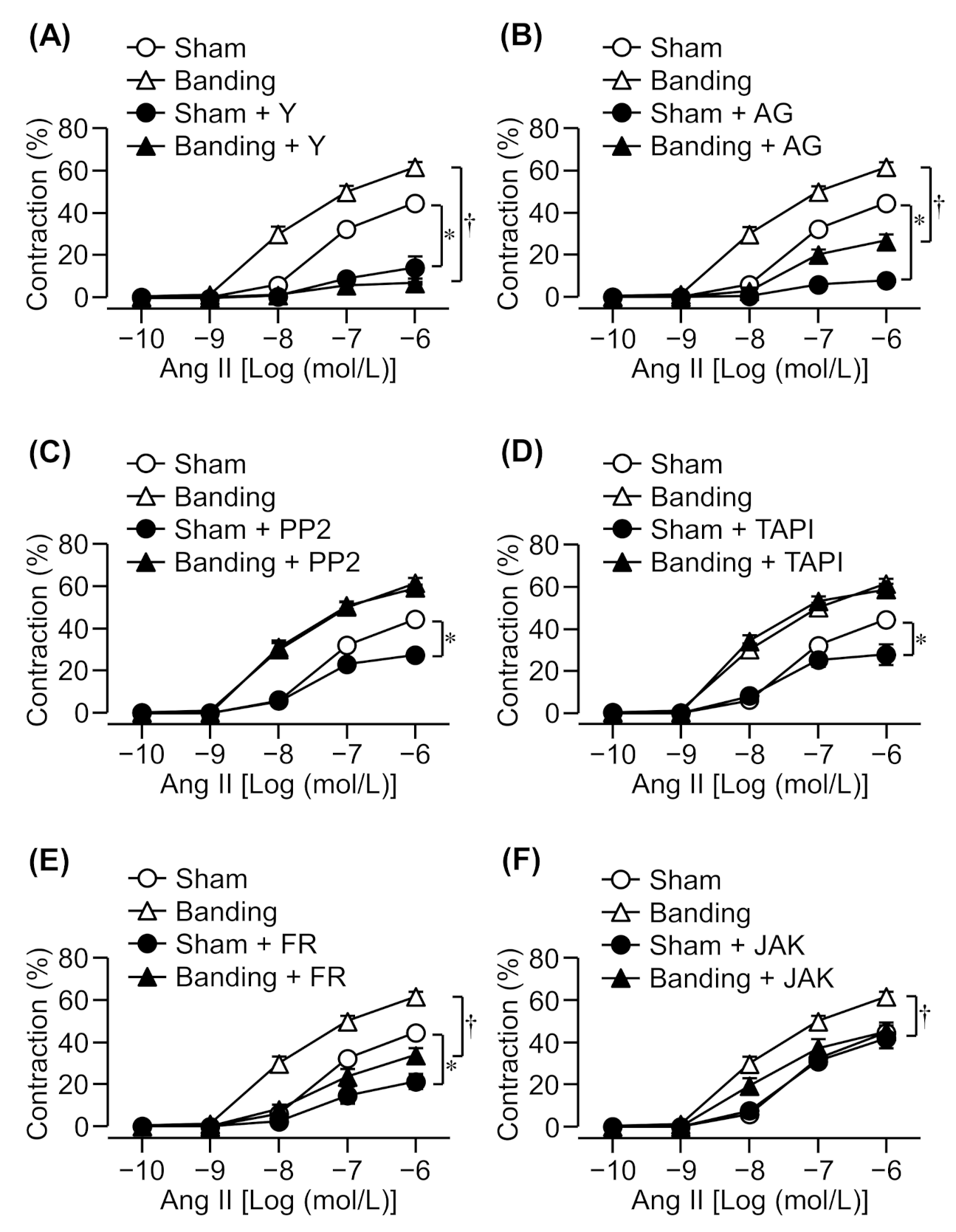
3.2. Effect of a Rho Kinase Inhibitor on Ang II-Induced Contraction
3.3. Effect of EGFR, Src, or Metalloproteinase Inhibitors on Ang II-Induced Contraction
3.4. Effect of an Erk1/2 Inhibitor on Ang II-Induced Contraction
3.5. Effect of a JAK2 Inhibitor on Ang II-Induced Contraction
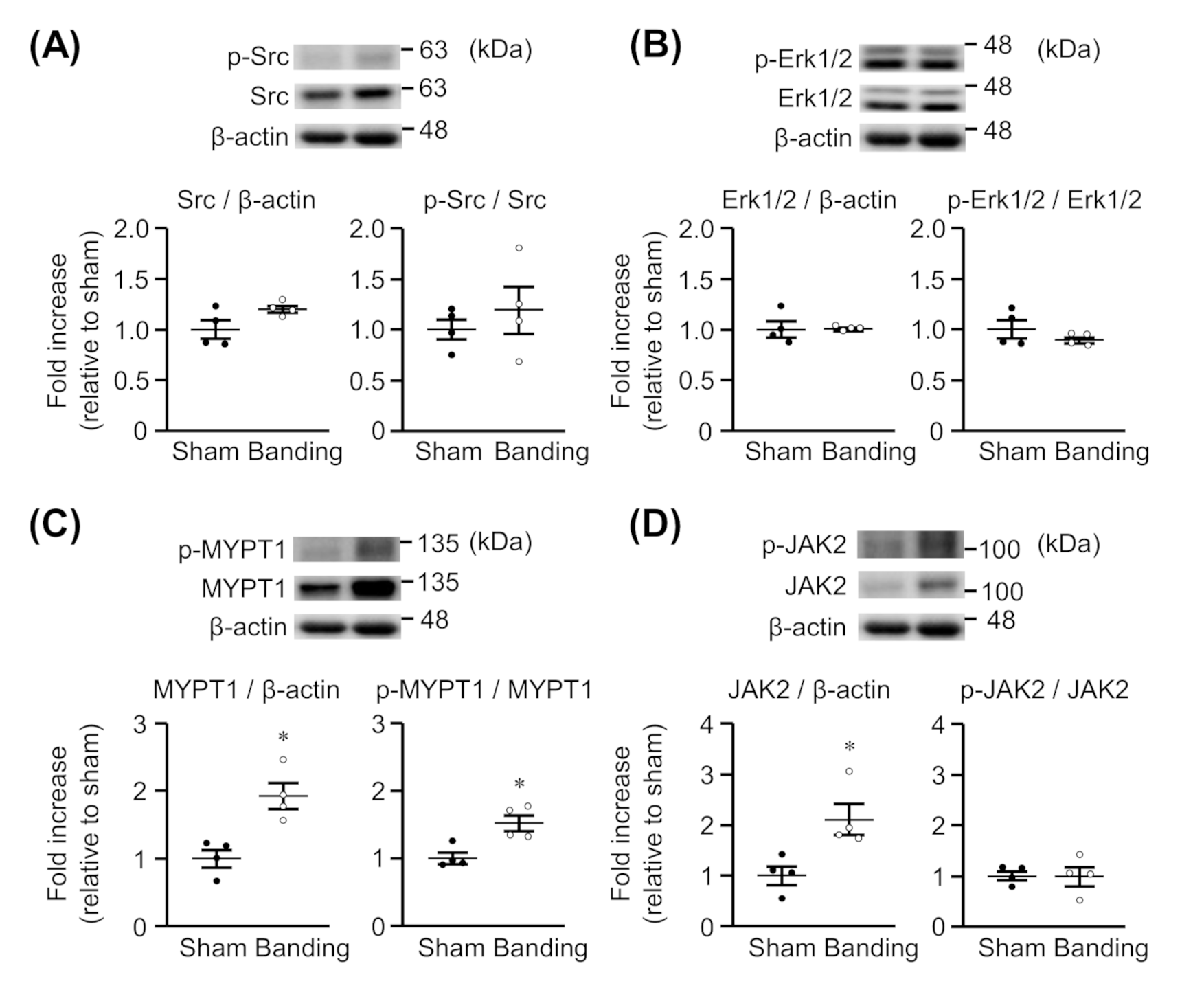
3.6. Expression and Phosphorylation of MYPT1, Src, Erk1/2, and JAK2 in Thoracic Aorta
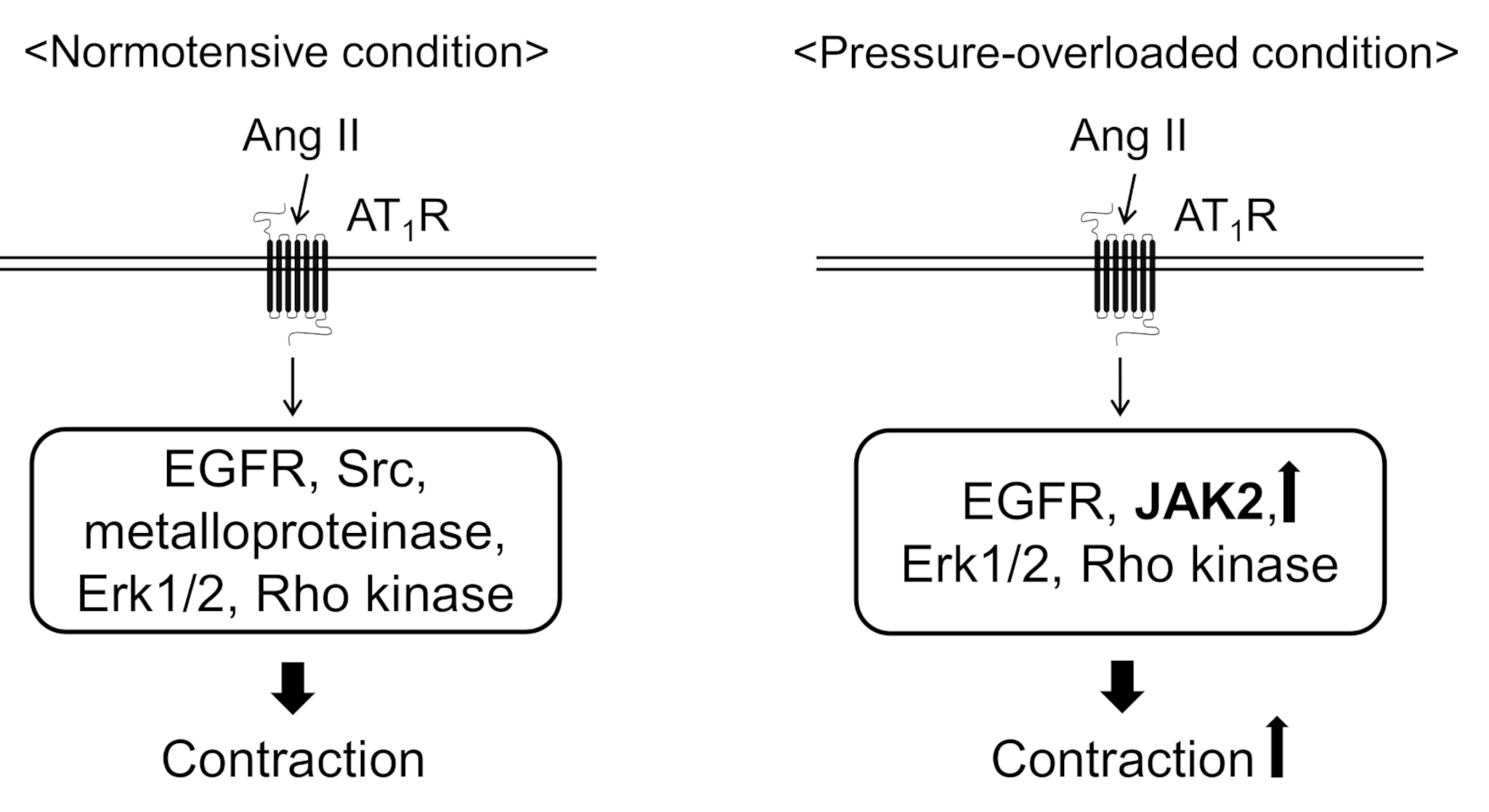
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Hirano, M.; Kanaide, H. Regulation of myosin phosphorylation and myofilament Ca2+ sensitivity in vascular smooth muscle. J. Smooth Muscle Res. 2004, 40, 219–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [Green Version]
- Sauro, M.D.; Sudakow, R.; Burns, S. In vivo effects of angiotensin II on vascular smooth muscle contraction and blood pressure are mediated through a protein tyrosine-kinase-dependent mechanism. J. Pharmacol. Exp. Ther. 1996, 277, 1744–1750. [Google Scholar] [PubMed]
- Touyz, R.M.; Berry, C. Recent advances in angiotensin II signaling. Braz. J. Med. Biol. Res. 2002, 35, 1001–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokemeyer, D.; Schmitz, U.; Kramer, H.J. Angiotensin II-induced growth of vascular smooth muscle cells requires an Src-dependent activation of the epidermal growth factor receptor. Kidney Int. 2000, 58, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Levesque, L.O.; Anand-Srivastava, M.B. Epidermal growth factor receptor transactivation by endogenous vasoactive peptides contributes to hyperproliferation of vascular smooth muscle cells of SHR. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1959–H1967. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Wu, X.H.; He, G.; Park, J.B.; Chen, X.; Vacher, J.; Rajapurohitam, V.; Schiffrin, E.L. Role of c-Src in the regulation of vascular contraction and Ca2+ signaling by angiotensin II in human vascular smooth muscle cells. J. Hypertens. 2001, 19, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhu, M.J.; Sreejayan, N.; Ren, J.; Du, M. Angiotensin II promotes smooth muscle cell proliferation and migration through release of heparin-binding epidermal growth factor and activation of EGF-receptor pathway. Mol. Cells 2005, 20, 263–270. [Google Scholar]
- Ishihata, A.; Tasaki, K.; Katano, Y. Involvement of p44/42 mitogen-activated protein kinases in regulating angiotensin II- and endothelin-1-induced contraction of rat thoracic aorta. Eur. J. Pharmacol. 2002, 445, 247–256. [Google Scholar] [CrossRef]
- Touyz, R.M.; el Mabrouk, M.; He, G.; Wu, X.H.; Schiffrin, E.L. Mitogen-activated protein/extracellular signal-regulated kinase inhibition attenuates angiotensin II-mediated signaling and contraction in spontaneously hypertensive rat vascular smooth muscle cells. Circ. Res. 1999, 84, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Biscardi, J.S.; Maa, M.C.; Tice, D.A.; Cox, M.E.; Leu, T.H.; Parsons, S.J. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 1999, 274, 8335–8343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Sato, A.; Aoto, M.; Fukami, Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun. 1995, 215, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Dempsey, P.J.; Frank, G.D.; Brailoiu, E.; Higuchi, S.; Suzuki, H.; Nakashima, H.; Eguchi, K.; Eguchi, S. Adam17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 2006, 26, e133–e137. [Google Scholar] [CrossRef]
- Guilluy, C.; Bregeon, J.; Toumaniantz, G.; Rolli-Derkinderen, M.; Retailleau, K.; Loufrani, L.; Henrion, D.; Scalbert, E.; Bril, A.; Torres, R.M.; et al. The Rho exchange factor arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 2010, 16, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yayama, K.; Horii, M.; Hiyoshi, H.; Takano, M.; Okamoto, H.; Kagota, S.; Kunitomo, M. Up-regulation of angiotensin II type 2 receptor in rat thoracic aorta by pressure-overload. J. Pharmacol. Exp. Ther. 2004, 308, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Matsuzaki, M.; Sasahara, T.; Shin, M.; Yayama, K. Orthovanadate-induced vasoconstriction of rat mesenteric arteries is mediated by Rho kinase-dependent inhibition of myosin light chain phosphatase. Biol. Pharm. Bull. 2015, 38, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Terada, Y.; Higashi, N.; Hidaka, Y.; Isomoto, Y.; Yayama, K. Protein tyrosine phosphatase inhibitor, orthovanadate, induces contraction via Rho kinase activation in mouse thoracic aortas. Biol. Pharm. Bull. 2019, 42, 877–885. [Google Scholar] [CrossRef]
- Yayama, K.; Sasahara, T.; Ohba, H.; Funasaka, A.; Okamoto, H. Orthovanadate-induced vasocontraction is mediated by the activation of Rho-kinase through Src-dependent transactivation of epidermal growth factor receptor. Pharmacol. Res. Perspect. 2014, 2, e00039. [Google Scholar] [CrossRef]
- Matrougui, K.; Tanko, L.B.; Loufrani, L.; Gorny, D.; Levy, B.I.; Tedgui, A.; Henrion, D. Involvement of Rho-kinase and the actin filament network in angiotensin II-induced contraction and extracellular signal-regulated kinase activity in intact rat mesenteric resistance arteries. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1288–1293. [Google Scholar] [CrossRef]
- Cox, R.H.; Lozinskaya, I.M. Augmented calcium currents in mesenteric artery branches of the spontaneously hypertensive rat. Hypertension 1995, 26, 1060–1064. [Google Scholar] [CrossRef]
- Hilgers, R.H.; Todd, J., Jr.; Webb, R.C. Increased PDZ-RhoGEF/RhoA/Rho kinase signaling in small mesenteric arteries of angiotensin II-induced hypertensive rats. J. Hypertens. 2007, 25, 1687–1697. [Google Scholar] [CrossRef]
- Ohtsu, H.; Suzuki, H.; Nakashima, H.; Dhobale, S.; Frank, G.D.; Motley, E.D.; Eguchi, S. Angiotensin II signal transduction through small GTP-binding proteins: Mechanism and significance in vascular smooth muscle cells. Hypertension 2006, 48, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.L.; Bregeon, J.; Devos, N.; Chadeuf, G.; Blanchard, A.; Azizi, M.; Pacaud, P.; Jeunemaitre, X.; Loirand, G. Angiotensin II activates the RhoA exchange factor Arhgef1 in humans. Hypertension 2015, 65, 1273–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crestani, S.; Webb, R.C.; da Silva-Santos, J.E. High-salt intake augments the activity of the RhoA/ROCK pathway and reduces intracellular calcium in arteries from rats. Am. J. Hypertens. 2017, 30, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Moriki, N.; Ito, M.; Seko, T.; Kureishi, Y.; Okamoto, R.; Nakakuki, T.; Kongo, M.; Isaka, N.; Kaibuchi, K.; Nakano, T. RhoA activation in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. Hypertens. Res. 2004, 27, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seko, T.; Ito, M.; Kureishi, Y.; Okamoto, R.; Moriki, N.; Onishi, K.; Isaka, N.; Hartshorne, D.J.; Nakano, T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ. Res. 2003, 92, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calo, L.A.; Davis, P.A.; Pagnin, E.; Maso, L.D.; Maiolino, G.; Seccia, T.M.; Pessina, A.C.; Rossi, G.P. Increased level of p63RhoGEF and RhoA/Rho kinase activity in hypertensive patients. J. Hypertens. 2014, 32, 331–338. [Google Scholar] [CrossRef]
- Kagiyama, S.; Eguchi, S.; Frank, G.D.; Inagami, T.; Zhang, Y.C.; Phillips, M.I. Angiotensin II-induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation 2002, 106, 909–912. [Google Scholar] [CrossRef]
- Schreier, B.; Hunerberg, M.; Rabe, S.; Mildenberger, S.; Bethmann, D.; Heise, C.; Sibilia, M.; Offermanns, S.; Gekle, M. Consequences of postnatal vascular smooth muscle EGFR deletion on acute angiotensin II action. Clin. Sci. 2016, 130, 19–33. [Google Scholar] [CrossRef]
- Touyz, R.M.; Wu, X.H.; He, G.; Salomon, S.; Schiffrin, E.L. Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased c-terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension 2002, 39, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, E.M.; Ma, X.; He, K.; Frank, S.J.; Ostrov, D.A.; Sayeski, P.P. Identification of 1,2,3,4,5,6-hexabromocyclohexane as a small molecule inhibitor of Jak2 tyrosine kinase autophosphorylation [correction of autophophorylation]. J. Med. Chem. 2005, 48, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Kirabo, A.; Oh, S.P.; Kasahara, H.; Wagner, K.U.; Sayeski, P.P. Vascular smooth muscle Jak2 deletion prevents angiotensin II-mediated neointima formation following injury in mice. J. Mol. Cell. Cardiol. 2011, 50, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, Y.; Shimokawa, H.; Matoba, T.; Kandabashi, T.; Satoh, S.; Hiroki, J.; Kaibuchi, K.; Takeshita, A. Involvement of Rho-kinase in hypertensive vascular disease: A novel therapeutic target in hypertension. FASEB J. 2001, 15, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, A.; Hirooka, Y.; Shimokawa, H.; Hironaga, K.; Setoguchi, S.; Takeshita, A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 2001, 38, 1307–1310. [Google Scholar] [CrossRef] [Green Version]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997, 389, 990–994. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Product Name | Chemical Name | Concentration Used (µM) |
|---|---|---|
| AG1478 | 4-(3-chloroanilino)-6,7-dimethoxyquinazoline | 10 |
| FR180204 | 5-(2-phenyl-pyrazolo[1,5-a]pyridin-3-yl)-1h-pyrazolo[3,4-c]pyridazin-3-ylamine | 10 |
| JAK2 Inhibitor II | 1,2,3,4,5,6-hexabromocyclohexane | 10 |
| PP2 | 4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1h-pyrazolo[3,4-d]pyrimidine | 3 |
| TAPI-0 | n-(R)-(2-[hydroxyaminocarbonyl]methyl)-4-methylpentanoyl-l-naphthylalanyl-l-alanine amide | 10 |
| Y-27632 | R-(+)-trans-n-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide | 10 |
| Antibody | Dilution | Catalog No. | Purchased from |
|---|---|---|---|
| MYPT1 | 1:600 | #2634 | Cell Signaling Technology (Danvers, MA, USA) |
| Thr-853-phosphorylated MYPT1 | 1:250 | CSB-PA020015 | CUSABIO (Houston, TX, USA) |
| Erk1/2 | 1:5000 | #4695 | Cell Signaling Technology (Danvers, MA, USA) |
| Thr-202/Tyr-204-phosphorylated Erk1/2 | 1:5000 | #9101 | Cell Signaling Technology (Danvers, MA, USA) |
| JAK2 | 1:500 | #3230 | Cell Signaling Technology (Danvers, MA, USA) |
| Tyr-1008-phosphorylated JAK2 | 1:200 | #8082 | Cell Signaling Technology (Danvers, MA, USA) |
| Src | 1:750 | #2102 | Cell Signaling Technology (Danvers, MA, USA) |
| Tyr-416-phosphorylated Src | 1:750 | #2101 | Cell Signaling Technology (Danvers, MA, USA) |
| β-actin | 1:5000 | A5441 | Sigma-Aldrich (St. Louis, MO, USA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terada, Y.; Yayama, K. Angiotensin II-Induced Vasoconstriction via Rho Kinase Activation in Pressure-Overloaded Rat Thoracic Aortas. Biomolecules 2021, 11, 1076. https://doi.org/10.3390/biom11081076
Terada Y, Yayama K. Angiotensin II-Induced Vasoconstriction via Rho Kinase Activation in Pressure-Overloaded Rat Thoracic Aortas. Biomolecules. 2021; 11(8):1076. https://doi.org/10.3390/biom11081076
Chicago/Turabian StyleTerada, Yuka, and Katsutoshi Yayama. 2021. "Angiotensin II-Induced Vasoconstriction via Rho Kinase Activation in Pressure-Overloaded Rat Thoracic Aortas" Biomolecules 11, no. 8: 1076. https://doi.org/10.3390/biom11081076
APA StyleTerada, Y., & Yayama, K. (2021). Angiotensin II-Induced Vasoconstriction via Rho Kinase Activation in Pressure-Overloaded Rat Thoracic Aortas. Biomolecules, 11(8), 1076. https://doi.org/10.3390/biom11081076