
The Labile Iron Pool Reacts Rapidly and Catalytically with Peroxynitrite


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Treatment
2.3. Quantification of the LIP
2.4. Fluorescence Experiments
2.5. In the Rate of Fluorescence Increase
2.6. Statistical Analysis
3. Results
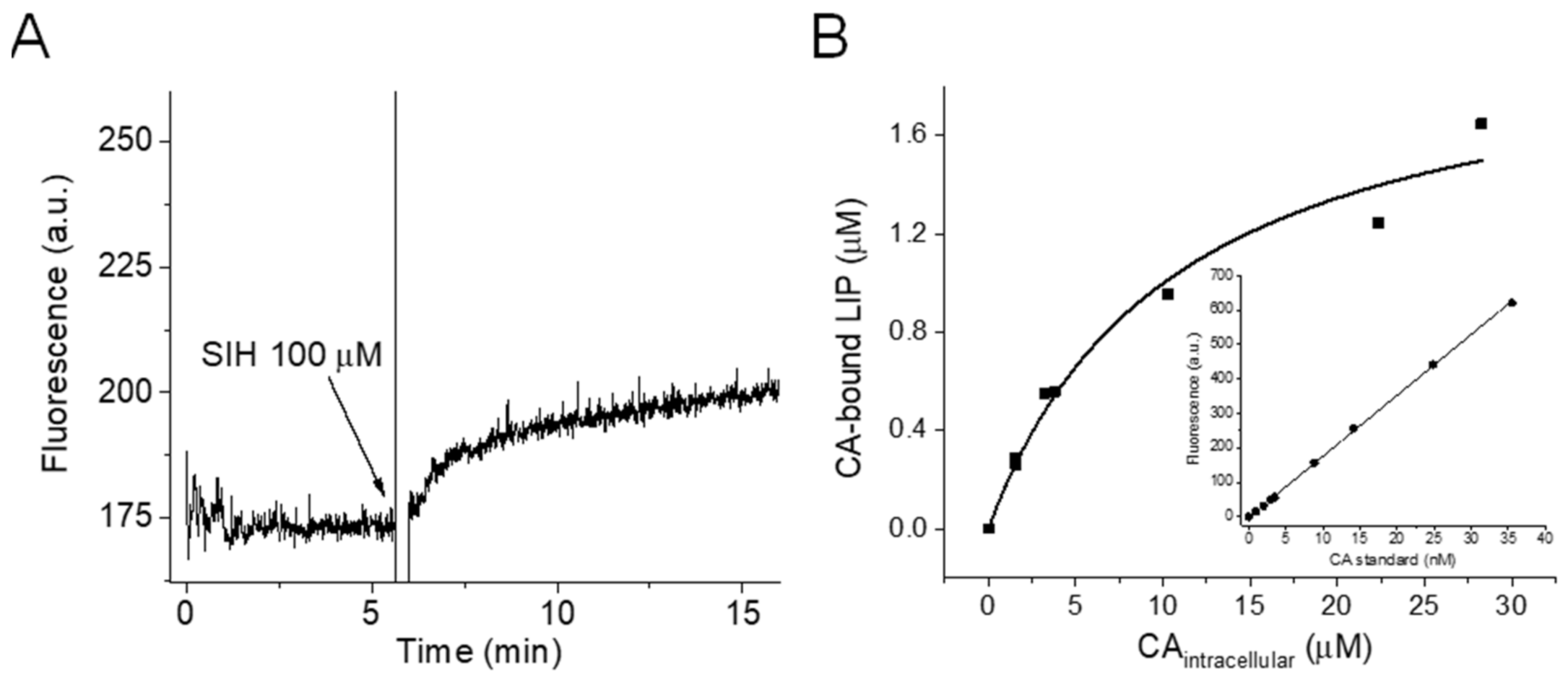
3.1. Quantifying the LIP
3.2. Checking Formation of Peroxynitrite
3.3. Monitoring Peroxynitrite-Dependent Oxidation
3.4. H2DCF Efficiently Detected Different Levels of in Cells Regardless of the Presence of the Chelator SIH
3.5. Removal of the LIP by Chelation Increases Peroxynitrite-Dependent Intracellular Oxidation
3.6. Catalytic Properties of the Reaction between the LIP and Peroxynitrite
3.7. The Rate Constant of the Reaction between the LIP and Peroxynitrite
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epsztejn, S.; Kakhlon, O.; Glickstein, H.; Breuer, W.; Cabantchik, Z.I. Fluorescence analysis of the labile iron pool of mammalian cells. Anal. Biochem. 1997, 248, 31–40. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Breuer, W.; Epsztejn, S.; Cabantchik, Z.I. Dynamics of the cytosolic chelatable iron pool of K562 cells. FEBS Lett. 1996, 382, 304–308. [Google Scholar] [CrossRef] [Green Version]
- Camarena, V.; Huff, T.C.; Wang, G.F. Epigenomic regulation by labile iron. Free Radic. Biol. Med. 2021, 170, 44–49. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res.-Fundam. Mol. Mech. Mutagenesis 2003, 531, 81–92. [Google Scholar] [CrossRef]
- Lin, L.S.; Wang, S.; Deng, H.Z.; Yang, W.J.; Rao, L.; Tian, R.; Liu, Y.; Yu, G.C.; Zhou, Z.J.; Song, J.B.; et al. Endogenous Labile Iron Pool-Mediated Free Radical Generation for Cancer Chemodynamic Therapy. J. Am. Chem. Soc. 2020, 142, 15320–15330. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Wang, Z.W.; Chen, L.; Zhang, C.; Liao, F.Y.; Wang, Y.W.; Wang, Y.; Luo, P.; Luo, M.; Shi, C.M. Artesunate induces ER-derived-ROS-mediated cell death by disrupting labile iron pool and iron redistribution in hepatocellular carcinoma cells. Am. J. Cancer Res. 2021, 11, 691. [Google Scholar]
- Golfeyz, S.; Lewis, S.; Weisberg, I.S. Hemochromatosis: Pathophysiology, evaluation, and management of hepatic iron overload with a focus on MRI. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 767–778. [Google Scholar] [CrossRef]
- Kew, M.C. Hepatic Iron Overload and Hepatocellular Carcinoma. Liver Cancer 2014, 3, 31–40. [Google Scholar] [CrossRef]
- Miyanishi, K.; Tanaka, S.; Sakamoto, H.; Kato, J. The role of iron in hepatic inflammation and hepatocellular carcinoma. Free Radic. Biol. Med. 2019, 133, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Shapiro, J.S.; Ardehali, H. Getting to the “Heart” of Cardiac Disease by Decreasing Mitochondrial Iron. Circ. Res. 2016, 119, 1164–1166. [Google Scholar] [CrossRef] [Green Version]
- Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Diagnosis and treatment of cardiac iron overload in transfusion-dependent thalassemia patients. Expert Rev. Hematol. 2018, 11, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Lawen, A.; Lane, D.J.R. Mammalian Iron Homeostasis in Health and Disease: Uptake, Storage, Transport, and Molecular Mechanisms of Action. Antioxid. Redox Signal. 2013, 18, 2473–2507. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Regulation of iron acquisition and storage: Consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81. [Google Scholar] [CrossRef]
- Moos, T.; Morgan, E.H. The metabolism of neuronal iron and its pathogenic role in neurological disease—Review. Redox-Act. Met. Neurol. Disord. 2004, 1012, 14–26. [Google Scholar] [CrossRef]
- Toledo, J.C.; Bosworth, C.A.; Hennon, S.W.; Mahtani, H.A.; Bergonia, H.A.; Lancaster, J.R. Nitric Oxide-induced Conversion of Cellular Chelatable Iron into Macromolecule-bound Paramagnetic Dinitrosyliron Complexes. J. Biol. Chem. 2008, 283, 28926–28933. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, C.Y.; Mahtani, H.K.; Du, J.; Patel, A.R.; Lancaster, J.R. Nitrosothiol Formation and Protection against Fenton Chemistry by Nitric Oxide-induced Dinitrosyliron Complex Formation from Anoxia-initiated Cellular Chelatable Iron Increase. J. Biol. Chem. 2014, 289, 19917–19927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickok, J.R.; Sahni, S.; Shen, H.; Arvind, A.; Antoniou, C.; Fung, L.W.M.; Thomas, D.D. Dinitrosyliron complexes are the most abundant nitric oxide-derived cellular adduct: Biological parameters of assembly and disappearance. Free Radic. Biol. Med. 2011, 51, 1558–1566. [Google Scholar] [CrossRef] [Green Version]
- Meczynska, S.; Lewandowska, H.; Sochanowicz, B.; Sadlo, J.; Kruszewski, M. Variable inhibitory effects on the formation of dinitrosyl iron complexes by deferoxamine and salicylaldehyde isonicotinoyl hydrazone in K562 cells. Hemoglobin 2008, 32, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Bosworth, C.A.; Toledo, J.C.; Zmijewski, J.W.; Li, Q.; Lancaster, J.R. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc. Natl. Acad. Sci. USA 2009, 106, 4671–4676. [Google Scholar] [CrossRef] [Green Version]
- Truzzi, D.R.; Augusto, O.; Ford, P.C. Thiyl radicals are co-products of dinitrosyl iron complex (DNIC) formation. Chem. Commun. 2019, 55, 9156–9159. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, F.C.; Condeles, A.L.; Lopes, A.K.B.; Facci, R.R.; Linares, E.; Truzzi, D.R.; Augusto, O.; Toledo, J.C. The labile iron pool attenuates peroxynitrite-dependent damage and can no longer be considered solely a pro-oxidative cellular iron source. J. Biol. Chem. 2018, 293, 8530–8542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997, 10, 1285–1292. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite—Implications for endothelial injury from nitric-oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [Green Version]
- Bonini, M.G.; Radi, R.; Ferrer-Sueta, G.; Ferreira, A.M.D.; Augusto, O. Direct EPR detection of the carbonate radical anion produced from peroxynitrite and carbon dioxide. J. Biol. Chem. 1999, 274, 10802–10806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Shimanovich, R.; Groves, J.T. Mechanisms of peroxynitrite decomposition catalyzed by FeTMPS, a bioactive sulfonated iron porphyrin. Arch. Biochem. Biophys. 2001, 387, 307–317. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Batinic-Haberle, I.; Spasojevic, I.; Fridovich, I.; Radi, R. Catalytic scavenging of peroxynitrite by isomeric Mn(III) N-methylpyridylporphyrins in the presence of reductants. Chem. Res. Toxicol. 1999, 12, 442–449. [Google Scholar] [CrossRef]
- Furtmuller, P.G.; Jantschko, W.; Zederbauer, M.; Schwanninger, M.; Jakopitsch, C.; Herold, S.; Koppenol, W.H.; Obinger, C. Peroxynitrite efficiently mediates the interconversion of redox intermediates of myeloperoxidase. Biochem. Biophys. Res. Commun. 2005, 337, 944–954. [Google Scholar] [CrossRef]
- Exner, M.; Herold, S. Kinetic and mechanistic studies of the peroxynitrite-mediated oxidation of oxymyoglobin and oxyhemoglobin. Chem. Res. Toxicol. 2000, 13, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Exner, M.; Boccini, F. The mechanism of the peroxynitrite-mediated oxidation of myoglobin in the absence and presence of carbon dioxide. Chem. Res. Toxicol. 2003, 16, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.K.; Das, J.V.; Gupta, V.K. A sensitive determination of paraquat by spectrophotometry. Talanta 1997, 45, 343–348. [Google Scholar] [CrossRef]
- Calderbank, A.; Yuen, S.H. An Ion-Exchange Method for Determining Paraquat Residues in Food Crops. Analyst 1965, 90, 99–106. [Google Scholar] [CrossRef]
- Bakalar, M.H.; Joffe, A.M.; Schmid, E.M.; Son, S.; Podolski, M.; Fletcher, D.A. Size-Dependent Segregation Controls Macrophage Phagocytosis of Antibody-Opsonized Targets. Cell 2018, 174, 131–142.e13. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lancaster, J.R. Calibration of nitric oxide flux generation from diazeniumdiolate (NO)-N-center dot donors. Nitric Oxide-Biol. Chem. 2009, 21, 69–75. [Google Scholar] [CrossRef]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Alvarez, M.N.; Trujillo, M.; Radi, R. Peroxynitrite formation from biochemical and cellular fluxes of nitric oxide and superoxide. Methods Enzymol. 2002, 359, 353–366. [Google Scholar]
- Zielonka, J.; Sikora, A.; Joseph, J.; Kalyanaraman, B. Peroxynitrite Is the Major Species Formed from Different Flux Ratios of Co-generated Nitric Oxide and Superoxide: Direct reaction with boronate-based fluorescent probe. J. Biol. Chem. 2010, 285, 14210–14216. [Google Scholar] [CrossRef] [Green Version]
- Wrona, M.; Patel, K.; Wardman, P. Reactivity of 2′,7′-dichlorodihydrofluorescein and dihydrorhodamine 123 and their oxidized forms toward carbonated nitrogen dioxide, and hydroxyl radicals. Free Radic. Biol. Med. 2005, 38, 262–270. [Google Scholar] [CrossRef]
- Masumoto, K.; Kissner, R.; Koppenol, W.H.; Sies, H. Kinetic study of the reaction of ebselen with peroxynitrite. FEBS Lett. 1996, 398, 179–182. [Google Scholar] [CrossRef] [Green Version]
- Possel, H.; Noack, H.; Augustin, W.; Keilhoff, G.; Wolf, G. 2,7-Dihydrodichlorofluorescein diacetate as a fluorescent marker for peroxynitrite formation. FEBS Lett. 1997, 416, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Wardman, P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: Progress, pitfalls, and prospects. Free Radic. Biol. Med. 2007, 43, 995–1022. [Google Scholar] [CrossRef]
- Sikora, A.; Zielonka, J.; Joseph, J.; Kalyanaraman, B. Oxidation of Coumarin Boronate to Hydroxycoumarin by Different Fluxes of Nitric Oxide and Superoxide: Quantitative Measurements of Peroxynitrite under Various Nitric Oxide Superoxide Fluxes. Free Radic. Biol. Med. 2009, 47, S36. [Google Scholar]
- Sikora, A.; Zielonka, J.; Lopez, M.; Dybala-Defratyka, A.; Joseph, J.; Marcinek, A.; Kalyanaraman, B. Reaction between Peroxynitrite and Boronates: EPR Spin-Trapping, HPLC Analyses, and Quantum Mechanical Study of the Free Radical Pathway. Chem. Res. Toxicol. 2011, 24, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Rios, N.; Piacenza, L.; Trujillo, M.; Martinez, A.; Demicheli, V.; Prolo, C.; Alvarez, M.N.; Lopez, G.V.; Radi, R. Sensitive detection and estimation of cell-derived peroxynitrite fluxes using fluorescein-boronate. Free Radic. Biol. Med. 2016, 101, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, G.L.; Postlethwait, E.M. On the hydrophobicity of nitrogen dioxide: Could there be a "lens" effect for NO2 reaction kinetics? Nitric Oxide-Biol. Chem. 2009, 21, 104–109. [Google Scholar] [CrossRef] [Green Version]
- Signorelli, S.; Moller, M.N.; Coitino, E.L.; Denicola, A. Nitrogen dioxide solubility and permeation in lipid membranes. Arch. Biochem. Biophys. 2011, 512, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Yoshida, M.; Miyamoto, Y.; Sato, K.; Kohno, M.; Sasamoto, K.; Miyazaki, K.; Ueda, S.; Maeda, H. Antagonistic action of Imidazolineoxyl N-oxides against endothelium-derived relaxing factor No through a radical reaction. Biochemistry 1993, 32, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Maeda, H. Direct quantitation of nitric oxide released from cells using liposome-encapsulated PTIO. In Proceedings of the 4th International Meeting on the Biology of Nitric Oxide, Amelia Island, FL, USA, September 1995; p. 171. [Google Scholar]
- Goldstein, S.; Russo, A.; Samuni, A. Reactions of PTIO and carboxy-PTIO with (NO)-N-center dot, (NO2)-N-center dot, and O-2(center dot). J. Biol. Chem. 2003, 278, 50949–50955. [Google Scholar] [CrossRef] [Green Version]
- Espey, M.G.; Thomas, D.D.; Miranda, K.M.; Wink, D.A. Focusing of nitric oxide mediated nitrosation and oxidative nitrosylation as a consequence of reaction with superoxide. Proc. Natl. Acad. Sci. USA 2002, 99, 11127–11132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, H.; Nakatsubo, N.; Kikuchi, K.; Kawahara, S.; Kirino, Y.; Nagoshi, H.; Hirata, Y.; Nagano, T. Detection and imaging of nitric oxide with novel fluorescent indicators: Diaminofluoresceins. Anal. Chem. 1998, 70, 2446–2453. [Google Scholar] [CrossRef] [PubMed]
- Espey, M.G.; Miranda, K.M.; Thomas, D.D.; Wink, D.A. Distinction between nitrosating mechanisms within human cells and aqueous solution. J. Biol. Chem. 2001, 276, 30085–30091. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.; Czapski, G. Indirect oxidation of ferrocyanide by peroxynitrite—Evidence against the formation of hydroxyl radicals. Nitric Oxide-Biol. Chem. 1997, 1, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Forni, L.G.; Moraarellano, V.O.; Packer, J.E.; Willson, R.L. Nitrogen-dioxide and related free-radicals—electron-transfer reactions with organic-compounds in solutions containing nitrite or nitrate. J. Chem. Soc.-Perkin Trans. 2 1986, 1–6. [Google Scholar] [CrossRef]
- Kirsch, M.; de Groot, H. Ascorbate is a potent antioxidant against peroxynitrite-induced oxidation reactions—Evidence that ascorbate acts by re-reducing substrate radicals produced by peroxynitrite. J. Biol. Chem. 2000, 275, 16702–16708. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.L.; Kong, X.L.; Xie, Y.Y.; Hider, R.C. The interaction of pyridoxal isonicotinoyl hydrazone (PIH) and salicylaldehyde isonicotinoyl hydrazone (SIH) with iron. J. Inorg. Biochem. 2018, 180, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Herold, S.; Koppenol, W.H. Peroxynitritometal complexes. Coord. Chem. Rev. 2005, 249, 499–506. [Google Scholar] [CrossRef]
- Damasceno, F.C.; Facci, R.R.; da Silva, T.M.; Toledo, J.C. Mechanisms and kinetic profiles of superoxide-stimulated nitrosative processes in cells using a diaminofluorescein probe. Free Radic. Biol. Med. 2014, 77, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Denicola, A.; Freeman, B.A.; Trujillo, M.; Radi, R. Peroxynitrite reaction with carbon dioxide/bicarbonate: Kinetics and influence on peroxynitrite-mediated oxidations. Arch. Biochem. Biophys. 1996, 333, 49–58. [Google Scholar] [CrossRef]
- Lymar, S.V.; Hurst, J.K. Rapid reaction between peroxonitrite ion and carbon-dioxide—implications for biological-activity. J. Am. Chem. Soc. 1995, 117, 8867–8868. [Google Scholar] [CrossRef]
- Ogusucu, R.; Rettori, D.; Munhoz, D.C.; Netto, L.E.S.; Augusto, O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: Rate constants by competitive kinetics. Free Radic. Biol. Med. 2007, 42, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.; Ferrer-Sueta, G.; Radi, R. Kinetic studies on peroxynitrite reduction by peroxiredoxins. In Nitric Oxide, Pt G: Oxidative and Nitrosative Stress in Redox Regulation of Cell Signaling; Cadenas, E., Packer, L., Eds.; Methods in Enzymology; Elsevier Academic Press Inc: San Diego, CA, USA, 2008; Volume 441, pp. 173–196. [Google Scholar]
- Yang, Y.S.; Balcarcel, R.R. Determination of carbon dioxide production rates for mammalian cells in 24-well plates. Biotechniques 2004, 36, 286–295. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors Affecting Protein Thiol Reactivity and Specificity in Peroxide Reduction. Chem. Res. Toxicol. 2011, 24, 434–450. [Google Scholar] [CrossRef]
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. Bioessays 2013, 35, 1050–1055. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.C.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamed, M.Y.; Silver, J. Studies on the reactions of ferric iron with glutathione and some related thiols II. Complex-formation in the pH range 3 to 7. Inorg. Chim. Acta-Bioinorg. Chem. 1983, 80, 115–122. [Google Scholar] [CrossRef]
- Hider, R.C.; Kong, X.L. Glutathione: A key component of the cytoplasmic labile iron pool. Biometals 2011, 24, 1179–1187. [Google Scholar] [CrossRef]
- Cho, E.A.; Song, H.K.; Lee, S.H.; Chung, B.H.; Lim, H.M.; Lee, M.K. Differential in vitro and cellular effects of iron chelators for hypoxia inducible factor hydroxylases. J. Cell. Biochem. 2013, 114, 864–873. [Google Scholar] [CrossRef]
- Philpott, C.C.; Patel, S.J.; Protchenko, O. Management versus miscues in the cytosolic labile iron pool: The varied functions of iron chaperones. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 9. [Google Scholar] [CrossRef]
- Patel, S.J.; Frey, A.G.; Palenchar, D.J.; Achar, S.; Bullough, K.Z.; Vashisht, A.; Wohlschlegel, J.A.; Philpott, C.C. A PCBP1-BolA2 chaperone complex delivers iron for cytosolic 2Fe-2S cluster assembly. Nat. Chem. Biol. 2019, 15, 872–881. [Google Scholar] [CrossRef]
- Shi, H.F.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [Green Version]
- Nandal, A.; Ruiz, J.C.; Subramanian, P.; Ghimire-Rijal, S.; Sinnamon, R.A.; Stemmler, T.L.; Bruick, R.K.; Philpott, C.C. Activation of the HIF Prolyl Hydroxylase by the Iron Chaperones PCBP1 and PCBP2. Cell Metab. 2011, 14, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.G.; Nandal, A.; Park, J.H.; Smith, P.M.; Yabe, T.; Ryu, M.S.; Ghosh, M.C.; Lee, J.; Rouault, T.A.; Park, M.H.; et al. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc. Natl. Acad. Sci. USA 2014, 111, 8031–8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leffers, H.; Dejgaard, K.; Celis, J.E. Characterization of 2 major cellular poly(rc)-binding human proteins, each containing 3 k-homologous (kh) domains. Eur. J. Biochem. 1995, 230, 447–453. [Google Scholar] [CrossRef]
- Zhou, L.F.; Zhang, L.X.; Wang, S.H.; Zhao, B.; Lv, H.H.; Shang, P. Labile iron affects pharmacological ascorbate-induced toxicity in osteosarcoma cell lines. Free Radic. Res. 2020, 54, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J. Biol. Chem. 2020, 295, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.P.; Liu, Z.X.; Zhang, Y.H.; Ma, L.; Song, E.Q.; Song, Y. “Iron free” zinc oxide nanoparticles with ion-leaking properties disrupt intracellular ROS and iron homeostasis to induce ferroptosis. Cell Death Dis. 2020, 11, 15. [Google Scholar] [CrossRef]
- Kakhlon, O.; Gruenbaum, Y.; Cabantchik, Z.L. Repression of ferritin expression increases the labile iron pool, oxidative stress, and short-term growth of human erythroleukemia cells. Blood 2001, 97, 2863–2871. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.C.M.; Iretskii, A.V.; Han, R.M.; Ford, P.C. Dinitrosyl Iron Complexes with Cysteine. Kinetics Studies of the Formation and Reactions of DNICs in Aqueous Solution. J. Am. Chem. Soc. 2015, 137, 328–336. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Condeles, A.L.; Toledo Junior, J.C. The Labile Iron Pool Reacts Rapidly and Catalytically with Peroxynitrite. Biomolecules 2021, 11, 1331. https://doi.org/10.3390/biom11091331
Condeles AL, Toledo Junior JC. The Labile Iron Pool Reacts Rapidly and Catalytically with Peroxynitrite. Biomolecules. 2021; 11(9):1331. https://doi.org/10.3390/biom11091331
Chicago/Turabian StyleCondeles, André Luís, and José Carlos Toledo Junior. 2021. "The Labile Iron Pool Reacts Rapidly and Catalytically with Peroxynitrite" Biomolecules 11, no. 9: 1331. https://doi.org/10.3390/biom11091331
APA StyleCondeles, A. L., & Toledo Junior, J. C. (2021). The Labile Iron Pool Reacts Rapidly and Catalytically with Peroxynitrite. Biomolecules, 11(9), 1331. https://doi.org/10.3390/biom11091331