
The Regulation of Rab GTPases by Phosphorylation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: The Potential Role of Phosphorylation in the Regulation of Rab
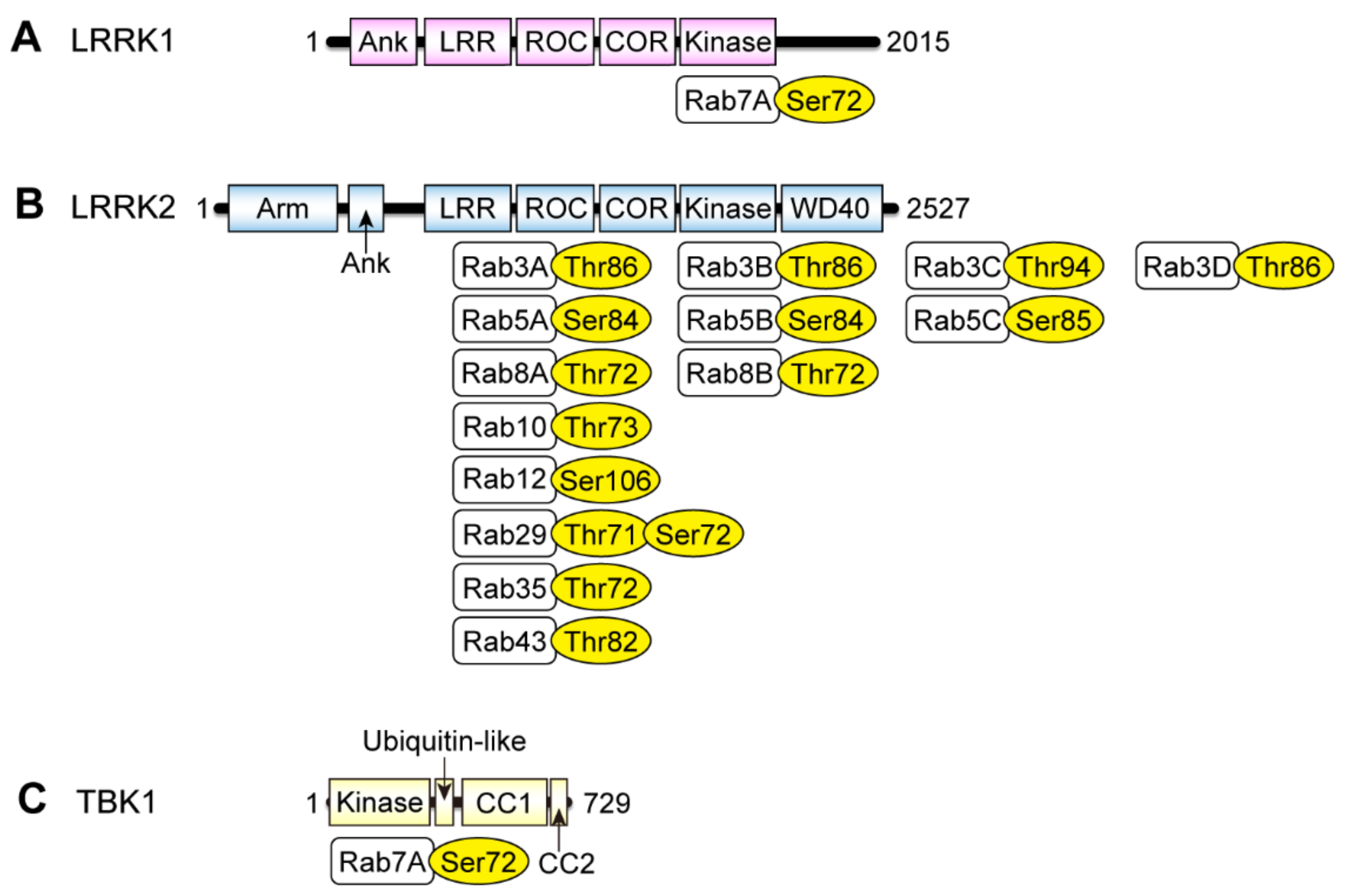
2. Rab Phosphorylation by LRRK2
2.1. Discovery of Rab Phosphorylation by LRRK2
2.2. Rab Phosphorylation by LRRK2 in Human Samples
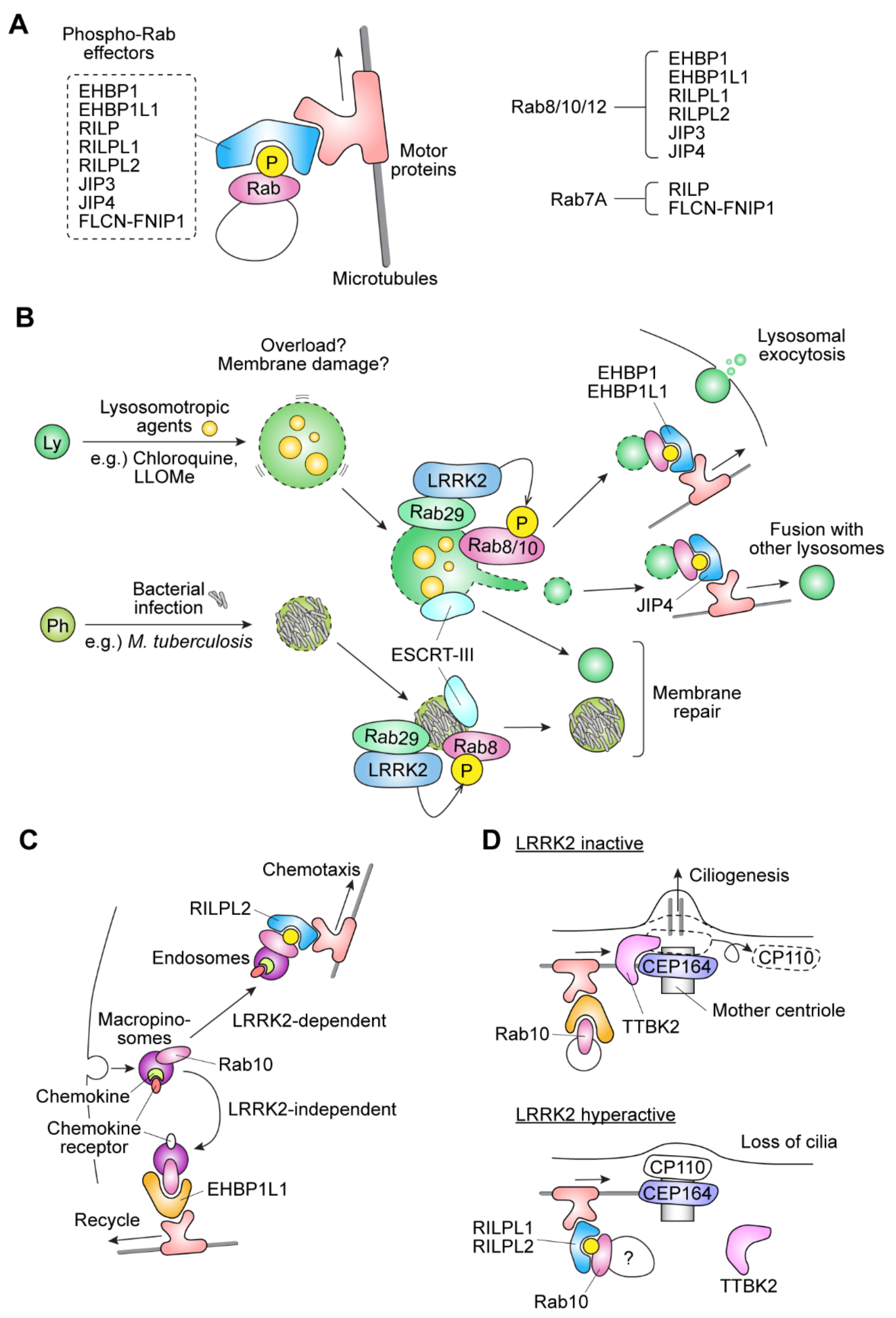
2.3. Functional Significance of Rab Phosphorylation by LRRK2
2.4. Potential Role of LRRK2-Rab in the Formation of Primary Cilia
2.5. Structural Evidence of the Effects of Rab Phosphorylation on the Effector Binding
2.6. Proposed Roles of LRRK2-Rab in α-Synuclein Propagation
2.7. Involvement of Rab29 in the LRRK2-Rab Pathway
2.8. Crosstalk with PINK1
3. Phosphorylation of Rab7A by LRRK1 and TBK1
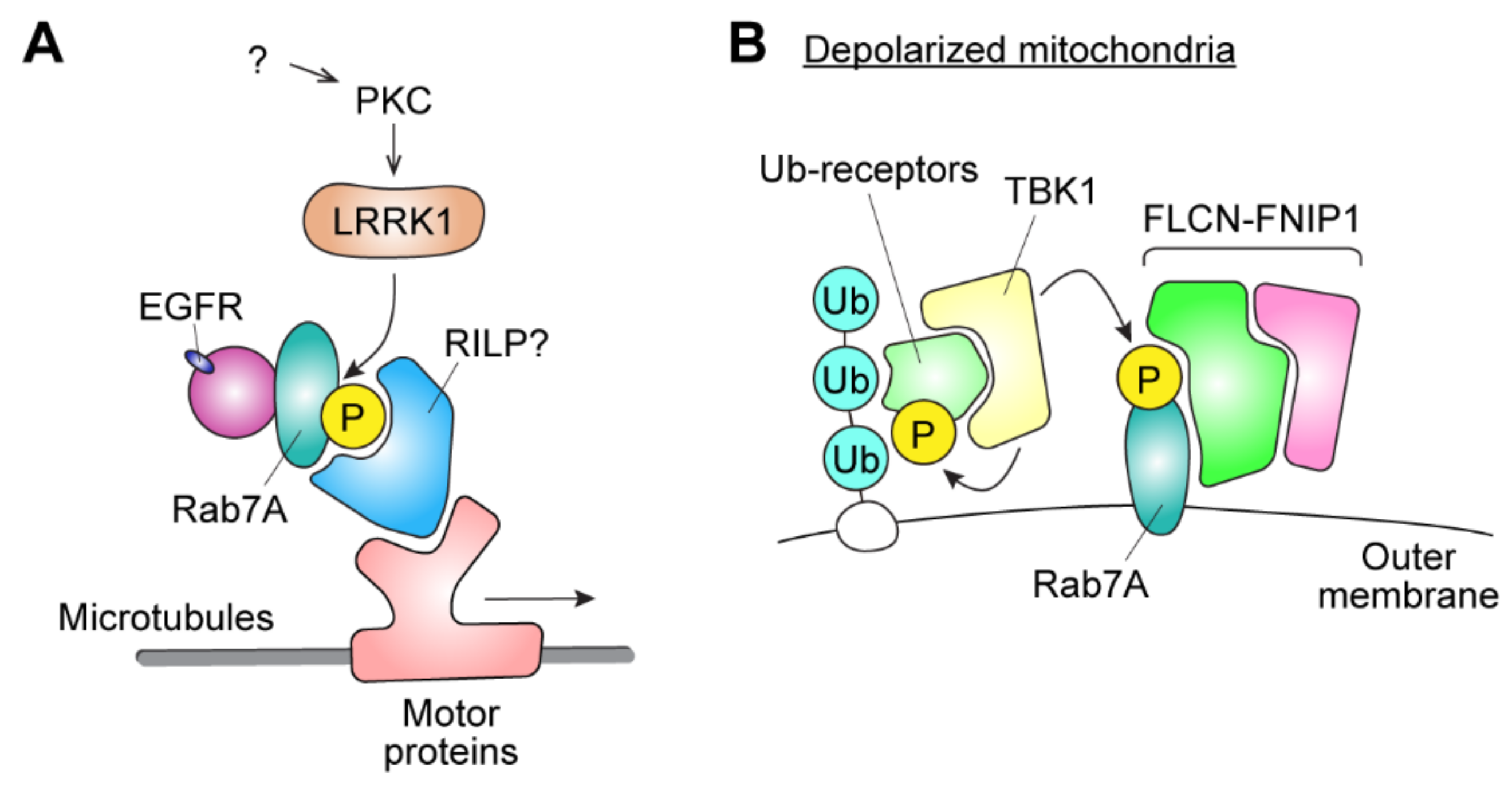
3.1. Rab7A Phosphorylation by LRRK1
3.2. Rab7A Phosphorylation by TBK1
3.3. The Possible Involvement of Rab7A Phosphorylation in Tumor Progression
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stenmark, H. Rab GTPases as Coordinators of Vesicle Traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Seabra, M.C.; Goldstein, J.L.; Südhof, T.C.; Brown, M.S. Rab Geranylgeranyl Transferase. A Multisubunit Enzyme That Prenylates GTP-Binding Proteins Terminating in Cys-X-Cys or Cys-Cys. J. Biol. Chem. 1992, 267, 14497–14503. [Google Scholar] [CrossRef]
- Seabra, M.C.; Brown, M.S.; Slaughter, C.A.; Südhof, T.C.; Goldstein, J.L. Purification of Component A of Rab Geranylgeranyl Transferase: Possible Identity with the Choroideremia Gene Product. Cell 1992, 70, 1049–1057. [Google Scholar] [CrossRef]
- Alexandrov, K.; Horiuchi, H.; Steele-Mortimer, O.; Seabra, M.C.; Zerial, M. Rab Escort Protein-1 Is a Multifunctional Protein That Accompanies Newly Prenylated Rab Proteins to Their Target Membranes. EMBO J. 1994, 13, 5262–5273. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-T.G.; Mishra, A.; Lambright, D.G. Structural Mechanisms for Regulation of Membrane Traffic by Rab GTPases. Traffic 2009, 10, 1377–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungar, D.; Hughson, F.M. SNARE Protein Structure and Function. Annu. Rev. Cell Dev. Biol. 2003, 19, 493–517. [Google Scholar] [CrossRef]
- Müller, M.P.; Goody, R.S. Molecular Control of Rab Activity by GEFs, GAPs and GDI. Small GTPases 2018, 9, 5–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.R.; Dirac-Svejstrup, A.B.; Soldati, T. Rab GDP Dissociation Inhibitor: Putting Rab GTPases in the Right Place. J. Biol. Chem. 1995, 270, 17057–17059. [Google Scholar] [CrossRef] [Green Version]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; de Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the Gene Containing Mutations That Cause PARK8-Linked Parkinson’s Disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.J.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Sveinbjornsdottir, S. The Clinical Symptoms of Parkinson’s Disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahn, S. Description of Parkinson’s Disease as a Clinical Syndrome. Ann. N. Y. Acad. Sci. 2003, 991, 1–14. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.-H.; Tomita, T.; Nakaya, K.; Lee, V.M.-Y.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of Alpha-Synuclein in Lewy Bodies of Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-Wide Association Study Identifies Common Variants at Four Loci as Genetic Risk Factors for Parkinson’s Disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-Wide Association Study Reveals Genetic Risk Underlying Parkinson’s Disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Araki, M.; Ito, G.; Tomita, T. Physiological and Pathological Functions of LRRK2: Implications from Substrate Proteins. Neuronal Signal. 2018, 2. [Google Scholar] [CrossRef] [Green Version]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s Disease-Associated Mutations in Leucine-Rich Repeat Kinase 2 Augment Kinase Activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics Reveals That Parkinson’s Disease Kinase LRRK2 Regulates a Subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, G.; Katsemonova, K.; Tonelli, F.; Lis, P.; Baptista, M.; Shpiro, N.; Duddy, G.; Wilson, S.; Ho, W.-L.; Ho, S.-L.; et al. Phos-Tag Analysis of Rab10 Phosphorylation by LRRK2: A Powerful Assay for Assessing Kinase Function and Inhibitors. Biochem. J. 2016, 473, 2671–2685. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic Proteomic Analysis of LRRK2-Mediated Rab GTPase Phosphorylation Establishes a Connection to Ciliogenesis. eLife 2017, 6, e31012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndsen, K.; Lis, P.; Yeshaw, W.M.; Wawro, P.S.; Nirujogi, R.S.; Wightman, M.; Macartney, T.; Dorward, M.; Knebel, A.; Tonelli, F.; et al. PPM1H Phosphatase Counteracts LRRK2 Signaling by Selectively Dephosphorylating Rab Proteins. eLife 2019, 8, e50416. [Google Scholar] [CrossRef]
- Jeong, G.R.; Jang, E.; Bae, J.R.; Jun, S.; Kang, H.C.; Park, C.; Shin, J.; Yamamoto, Y.; Tanaka-Yamamoto, K.; Dawson, V.L.; et al. Dysregulated Phosphorylation of Rab GTPases by LRRK2 Induces Neurodegeneration. Mol. Neurodegener. 2018, 13, 1–17. [Google Scholar] [CrossRef]
- Fujimoto, T.; Kuwahara, T.; Eguchi, T.; Sakurai, M.; Komori, T.; Iwatsubo, T. Parkinson’s Disease-Associated Mutant LRRK2 Phosphorylates Rab7L1 and Modifies Trans-Golgi Morphology. Biochem. Biophys. Res. Commun. 2018, 495, 1708–1715. [Google Scholar] [CrossRef]
- Thirstrup, K.; Dächsel, J.C.; Oppermann, F.S.; Williamson, D.S.; Smith, G.P.; Fog, K.; Christensen, K.V. Selective LRRK2 Kinase Inhibition Reduces Phosphorylation of Endogenous Rab10 and Rab12 in Human Peripheral Mononuclear Blood Cells. Sci. Rep. 2017, 7, 10300. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Howden, A.J.M.; Sarhan, A.R.; Lis, P.; Ito, G.; Martinez, T.N.; Brockmann, K.; Gasser, T.; Alessi, D.R.; Sammler, E.M. Interrogating Parkinson’s Disease LRRK2 Kinase Pathway Activity by Assessing Rab10 Phosphorylation in Human Neutrophils. Biochem. J. 2018, 475, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Atashrazm, F.; Hammond, D.; Perera, G.; Bolliger, M.F.; Matar, E.; Halliday, G.M.; Schüle, B.; Lewis, S.J.G.; Nichols, R.J.; Dzamko, N. LRRK2-Mediated Rab10 Phosphorylation in Immune Cells from Parkinson’s Disease Patients. Mov. Disord. 2019, 34, 406–415. [Google Scholar] [CrossRef]
- Karayel, Ö.; Tonelli, F.; Virreira Winter, S.; Geyer, P.E.; Fan, Y.; Sammler, E.M.; Alessi, D.R.; Steger, M.; Mann, M. Accurate MS-Based Rab10 Phosphorylation Stoichiometry Determination as Readout for LRRK2 Activity in Parkinson’s Disease. Mol. Cell. Proteomics 2020, 19, 1546–1560. [Google Scholar] [CrossRef]
- Fan, Y.; Nirujogi, R.S.; Garrido, A.; Ruiz-Martínez, J.; Bergareche-Yarza, A.; Mondragón-Rezola, E.; Vinagre-Aragón, A.; Croitoru, I.; Pagola, A.G.; Markinez, L.P.; et al. R1441G but Not G2019S Mutation Enhances LRRK2 Mediated Rab10 Phosphorylation in Human Peripheral Blood Neutrophils. Acta Neuropathol. 2021, 142, 475–494. [Google Scholar] [CrossRef]
- Wang, X.; Negrou, E.; Maloney, M.T.; Bondar, V.V.; Andrews, S.V.; Montalban, M.; Llapashtica, C.; Maciuca, R.; Nguyen, H.; Solanoy, H.; et al. Understanding LRRK2 Kinase Activity in Preclinical Models and Human Subjects through Quantitative Analysis of LRRK2 and PT73 Rab10. Sci. Rep. 2021, 11, 12900. [Google Scholar] [CrossRef]
- Maio, R.D.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; Miranda, B.R.D.; Zharikov, A.; Laar, A.V.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 Activation in Idiopathic Parkinson’s Disease. Sci. Transl. Med. 2018, 10, 5429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, T.; Kuwahara, T.; Sakurai, M.; Komori, T.; Fujimoto, T.; Ito, G.; Yoshimura, S.; Harada, A.; Fukuda, M.; Koike, M.; et al. LRRK2 and Its Substrate Rab GTPases Are Sequentially Targeted onto Stressed Lysosomes and Maintain Their Homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, e9115–e9124. [Google Scholar] [CrossRef] [Green Version]
- Waschbüsch, D.; Purlyte, E.; Pal, P.; McGrath, E.; Alessi, D.R.; Khan, A.R. Structural Basis for Rab8a Recruitment of RILPL2 via LRRK2 Phosphorylation of Switch 2. Structure 2020, 28, 406–417. [Google Scholar] [CrossRef]
- Heo, J.-M.; Ordureau, A.; Swarup, S.; Paulo, J.A.; Shen, K.; Sabatini, D.M.; Harper, J.W. RAB7A Phosphorylation by TBK1 Promotes Mitophagy via the PINK-PARKIN Pathway. Sci. Adv. 2018, 4, 443. [Google Scholar] [CrossRef] [Green Version]
- Hanafusa, H.; Yagi, T.; Ikeda, H.; Hisamoto, N.; Nishioka, T.; Kaibuchi, K.; Shirakabe, K.; Matsumoto, K. LRRK1 Phosphorylation of Rab7 at Ser-72 Links Trafficking of EGFR-Containing Endosomes to Its Effector RILP. J. Cell Sci. 2019, 132, 228809. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, T.; Funakawa, K.; Komori, T.; Sakurai, M.; Yoshii, G.; Eguchi, T.; Fukuda, M.; Iwatsubo, T. Roles of Lysosomotropic Agents on LRRK2 Activation and Rab10 Phosphorylation. Neurobiol. Dis. 2020, 145, 105081. [Google Scholar] [CrossRef]
- Lee, H.; Flynn, R.; Sharma, I.; Haberman, E.; Carling, P.J.; Nicholls, F.J.; Stegmann, M.; Vowles, J.; Haenseler, W.; Wade-Martins, R.; et al. LRRK2 Is Recruited to Phagosomes and Co-Recruits RAB8 and RAB10 in Human Pluripotent Stem Cell-Derived Macrophages. Stem Cell Rep. 2020, 14, 940–955. [Google Scholar] [CrossRef]
- Herbst, S.; Campbell, P.; Harvey, J.; Bernard, E.M.; Papayannopoulos, V.; Wood, N.W.; Morris, H.R.; Gutierrez, M.G. LRRK2 Activation Controls the Repair of Damaged Endomembranes in Macrophages. EMBO J. 2020, 39, e104494. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Beilina, A.; Williamson, C.D.; Lindberg, E.; Kluss, J.H.; Saez-Atienzar, S.; Landeck, N.; Kumaran, R.; Mamais, A.; Bleck, C.K.E.; et al. LRRK2 Mediates Tubulation and Vesicle Sorting from Lysosomes. Sci. Adv. 2020, 6, 2454. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, E.; Zhao, H.T.; Cole, T.; West, A.B. LRRK2 and Rab10 Coordinate Macropinocytosis to Mediate Immunological Responses in Phagocytes. EMBO J. 2020, 39, e104862. [Google Scholar] [CrossRef] [PubMed]
- Sobu, Y.; Wawro, P.S.; Dhekne, H.S.; Yeshaw, W.M.; Pfeffer, S.R. Pathogenic LRRK2 Regulates Ciliation Probability Upstream of Tau Tubulin Kinase 2 via Rab10 and RILPL1 Proteins. Proc. Natl. Acad. Sci. USA 2021, 118, e2005894118. [Google Scholar] [CrossRef] [PubMed]
- Dhekne, H.S.; Yanatori, I.; Gomez, R.C.; Tonelli, F.; Diez, F.; Schüle, B.; Steger, M.; Alessi, D.R.; Pfeffer, S.R. A Pathway for Parkinson’s Disease LRRK2 Kinase to Block Primary Cilia and Sonic Hedgehog Signaling in the Brain. eLife 2018, 7, e40202. [Google Scholar] [CrossRef] [PubMed]
- Schaub, J.R.; Stearns, T. The Rilp-like Proteins Rilpl1 and Rilpl2 Regulate Ciliary Membrane Content. Mol. Biol. Cell 2013, 24, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Dhekne, H.S.; Yanatori, I.; Vides, E.G.; Sobu, Y.; Diez, F.; Tonelli, F.; Pfeffer, S.R. LRRK2-Phosphorylated Rab10 Sequesters Myosin Va with RILPL2 during Ciliogenesis Blockade. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The Graded Response to Sonic Hedgehog Depends on Cilia Architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Threlfell, S.; Clements, M.A.; Khodai, T.; Pienaar, I.S.; Exley, R.; Wess, J.; Cragg, S.J. Striatal Muscarinic Receptors Promote Activity Dependence of Dopamine Transmission via Distinct Receptor Subtypes on Cholinergic Interneurons in Ventral versus Dorsal Striatum. J. Neurosci. 2010, 30, 3398–3408. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, L.E.; Verbitsky, M.; Blesa, J.; Jackson-Lewis, V.; Paredes, D.; Tillack, K.; Phani, S.; Kramer, E.R.; Przedborski, S.; Kottmann, A.H. Sonic Hedgehog Maintains Cellular and Neurochemical Homeostasis in the Adult Nigrostriatal Circuit. Neuron 2012, 75, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waschbüsch, D.; Purlyte, E.; Khan, A.R. Dual Arginine Recognition of LRRK2 Phosphorylated Rab GTPases. Biophys. J. 2021, 120, 1846–1855. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.; Brundin, P. Prion-like propagation of pathology in Parkinson disease. In Handbook of Clinical Neurology; Prion-Like Propagation of Pathology in Parkinson Disease; Elsevier: Amsterdam, The Netherlands, 2018; pp. 321–335. ISBN 978-0-444-63945-5. [Google Scholar]
- Braak, H.; Tredici, K.D.; Rüb, U.; Vos, R.A.I.D.; Steur, E.N.H.J.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Henderson, M.X.; Trojanowski, J.Q.; Lee, V.M.-Y. α-Synuclein Pathology in Parkinson’s Disease and Related α-Synucleinopathies. Neurosci. Lett. 2019, 709, 134316. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 Mutant IPSC-Derived DA Neurons Demonstrate Increased Susceptibility to Oxidative Stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Danés, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jiménez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-Specific Phenotypes in Dopamine Neurons from Human IPS-based Models of Genetic and Sporadic Parkinson’s Disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic Correction of a LRRK2 Mutation in Human IPSCs Links Parkinsonian Neurodegeneration to ERK-Dependent Changes in Gene Expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Keshiya, S.; Perera, G.; Schramko, L.; Halliday, G.M.; Dzamko, N. LRRK2 Kinase Inhibitors Reduce Alpha-Synuclein in Human Neuronal Cell Lines with the G2019S Mutation. Neurobiol. Dis. 2020, 144, 105049. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.L.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments α-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef]
- Daher, J.P.L.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of α-Synuclein-Mediated Dopaminergic Neurodegeneration in LRRK2-Deficient Rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, M.X.; Peng, C.; Trojanowski, J.Q.; Lee, V.M.Y. LRRK2 Activity Does Not Dramatically Alter α-Synuclein Pathology in Primary Neurons. Acta Neuropathol. Commun. 2018, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Sengupta, M.; McGeary, I.; Zhang, B.; Olufemi, M.F.; Brown, H.; Trojanowski, J.Q.; Lee, V.M.Y. LRRK2 Inhibition Does Not Impart Protection from α-Synuclein Pathology and Neuron Death in Non-Transgenic Mice. Acta Neuropathol. Commun. 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schüle, B.; et al. LRRK2 Modifies α-Syn Pathology and Spread in Mouse Models and Human Neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-J.; Kim, D.-K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 Kinase Regulates α-Synuclein Propagation via RAB35 Phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Bryant, N.; Kumaran, R.; Beilina, A.; Abeliovich, A.; Cookson, M.R.; West, A.B. LRRK2 Phosphorylates Membrane-Bound Rabs and Is Activated by GTP-Bound Rab7L1 to Promote Recruitment to the Trans-Golgi Network. Hum. Mol. Genet. 2018, 27, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Beilina, A.; Rudenko, I.N.; Kaganovich, A.; Civiero, L.; Chau, H.; Kalia, S.K.; Kalia, L.V.; Lobbestael, E.; Chia, R.; Ndukwe, K.; et al. Unbiased Screen for Interactors of Leucine-Rich Repeat Kinase 2 Supports a Common Pathway for Sporadic and Familial Parkinson Disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2626–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, R.; Haddock, S.; Beilina, A.; Rudenko, I.N.; Mamais, A.; Kaganovich, A.; Li, Y.; Kumaran, R.; Nalls, M.A.; Cookson, M.R. Phosphorylation of LRRK2 by Casein Kinase 1α Regulates Trans-Golgi Clustering via Differential Interaction with ARHGEF7. Nat. Commun. 2014, 5, 5827. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ma, Z.; Xu, X.; Wang, Z.; Sun, L.; Zhou, Y.; Lin, X.; Hong, W.; Wang, T. A Role of Rab29 in the Integrity of the Trans-Golgi Network and Retrograde Trafficking of Mannose-6-Phosphate Receptor. PLoS ONE 2014, 9, e96242. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, M.; Fukuda, M. Small GTPase Rab2B and Its Specific Binding Protein Golgi-Associated Rab2B Interactor-like 4 (GARI-L4) Regulate Golgi Morphology. J. Biol. Chem. 2015, 290, 22250–22261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, R.C.; Wawro, P.; Lis, P.; Alessi, D.R.; Pfeffer, S.R. Membrane Association but Not Identity Is Required for LRRK2 Activation and Phosphorylation of Rab GTPases. J. Cell Biol. 2019, 218, 4157–4170. [Google Scholar] [CrossRef] [Green Version]
- Kalogeropulou, A.F.; Freemantle, J.B.; Lis, P.; Vides, E.G.; Polinski, N.K.; Alessi, D.R. Endogenous Rab29 Does Not Impact Basal or Stimulated LRRK2 Pathway Activity. Biochem. J. 2020, 110, 1689. [Google Scholar] [CrossRef]
- Kluss, J.H.; Beilina, A.; Lewis, P.A.; Cookson, M.R.; Bonet-Ponce, L. Membrane Targeting Activates Leucine-Rich Repeat Kinase 2 with Differential Effects on Downstream Rab Activation. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.12.01.406223v2 (accessed on 9 September 2021). [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, 170–185. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-C.; Kondapalli, C.; Lehneck, R.; Procter, J.B.; Dill, B.D.; Woodroof, H.I.; Gourlay, R.; Peggie, M.; Macartney, T.J.; Corti, O.; et al. Phosphoproteomic Screening Identifies Rab GTPases as Novel Downstream Targets of PINK1. EMBO J. 2015, 34, 2840–2861. [Google Scholar] [CrossRef]
- Vieweg, S.; Mulholland, K.; Bräuning, B.; Kachariya, N.; Lai, Y.-C.; Toth, R.; Singh, P.K.; Volpi, I.; Sattler, M.; Groll, M.; et al. PINK1-Dependent Phosphorylation of Serine111 within the SF3 Motif of Rab GTPases Impairs Effector Interactions and LRRK2-Mediated Phosphorylation at Threonine72. Biochem. J. 2020, 477, 1651–1668. [Google Scholar] [CrossRef] [Green Version]
- Sejwal, K.; Chami, M.; Rémigy, H.; Vancraenenbroeck, R.; Sibran, W.; Sütterlin, R.; Baumgartner, P.; McLeod, R.; Chartier-Harlin, M.-C.; Baekelandt, V.; et al. Cryo-EM Analysis of Homodimeric Full-Length LRRK2 and LRRK1 Protein Complexes. Sci. Rep. 2017, 7, 8667. [Google Scholar] [CrossRef] [Green Version]
- Korr, D.; Toschi, L.; Donner, P.; Pohlenz, H.-D.; Kreft, B.; Weiss, B. LRRK1 Protein Kinase Activity Is Stimulated upon Binding of GTP to Its Roc Domain. Cell. Signal. 2006, 18, 910–920. [Google Scholar] [CrossRef]
- Ito, G.; Okai, T.; Fujino, G.; Takeda, K.; Ichijo, H.; Katada, T.; Iwatsubo, T. GTP Binding Is Essential to the Protein Kinase Activity of LRRK2, a Causative Gene Product for Familial Parkinson’s Disease. Biochemistry 2007, 46, 1380–1388. [Google Scholar] [CrossRef]
- Malik, A.U.; Karapetsas, A.; Nirujogi, R.S.; Mathea, S.; Chatterjee, D.; Pal, P.; Lis, P.; Taylor, M.; Purlyte, E.; Gourlay, R.; et al. Deciphering the LRRK Code: LRRK1 and LRRK2 Phosphorylate Distinct Rab Proteins and Are Regulated by Diverse Mechanisms. Biochem. J. 2021, 478, 553–578. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, H.; Kedashiro, S.; Tezuka, M.; Funatsu, M.; Usami, S.; Toyoshima, F.; Matsumoto, K. PLK1-Dependent Activation of LRRK1 Regulates Spindle Orientation by Phosphorylating CDK5RAP2. Nat. Cell Biol. 2015, 17, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, H.; Ishikawa, K.; Kedashiro, S.; Saigo, T.; Iemura, S.-I.; Natsume, T.; Komada, M.; Shibuya, H.; Nara, A.; Matsumoto, K. Leucine-Rich Repeat Kinase LRRK1 Regulates Endosomal Trafficking of the EGF Receptor. Nat. Commun. 2011, 2, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedashiro, S.; Pastuhov, S.I.; Nishioka, T.; Watanabe, T.; Kaibuchi, K.; Matsumoto, K.; Hanafusa, H. LRRK1-Phosphorylated CLIP-170 Regulates EGFR Trafficking by Recruiting P150Glued to Microtubule plus Ends. J. Cell Sci. 2015, 128, 829. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lin, J.; Han, J. Receptor-Interacting Protein (RIP) Kinase Family. Cell. Mol. Immunol. 2010, 7, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhang, C.; Li, M.; Zhao, C.; Zheng, Y. A System-Wide Spatiotemporal Characterization of ErbB Receptor Complexes by Subcellular Fractionation Integrated Quantitative Mass Spectrometry. Anal. Chem. 2021, 93, 7933–7941. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome Sequencing in Amyotrophic Lateral Sclerosis Identifies Risk Genes and Pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 Causes Familial ALS and Fronto-Temporal Dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Dzamko, N.; Inesta-Vaquera, F.; Zhang, J.; Xie, C.; Cai, H.; Arthur, J.S.C.; Tan, L.; Choi, H.G.; Gray, N.S.; Cohen, P.; et al. The IkappaB Kinase Family Phosphorylates the Parkinson’s Disease Kinase LRRK2 at Ser935 and Ser910 during Toll-like Receptor Signaling. PLoS ONE 2012, 7, e39132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, S.R.; Maddika, S. PTEN Modulates EGFR Late Endocytic Trafficking and Degradation by Dephosphorylating Rab7. Nat. Commun. 2016, 7, 10689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, J.L.; Zhu, Z.; Thai, T.C.; Mahadevan, N.R.; Mertins, P.; Knelson, E.H.; Piel, B.P.; Han, S.; Jaffe, J.D.; Carr, S.A.; et al. Phosphorylation of RAB7 by TBK1/IKKε Regulates Innate Immune Signaling in Triple-Negative Breast Cancer. Cancer Res. 2020, 80, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Nagai, Y.; Kajihara, Y.; Ito, G.; Tomita, T. The Regulation of Rab GTPases by Phosphorylation. Biomolecules 2021, 11, 1340. https://doi.org/10.3390/biom11091340
Xu L, Nagai Y, Kajihara Y, Ito G, Tomita T. The Regulation of Rab GTPases by Phosphorylation. Biomolecules. 2021; 11(9):1340. https://doi.org/10.3390/biom11091340
Chicago/Turabian StyleXu, Lejia, Yuki Nagai, Yotaro Kajihara, Genta Ito, and Taisuke Tomita. 2021. "The Regulation of Rab GTPases by Phosphorylation" Biomolecules 11, no. 9: 1340. https://doi.org/10.3390/biom11091340
APA StyleXu, L., Nagai, Y., Kajihara, Y., Ito, G., & Tomita, T. (2021). The Regulation of Rab GTPases by Phosphorylation. Biomolecules, 11(9), 1340. https://doi.org/10.3390/biom11091340