
Multi-Organ Crosstalk with Endocrine Pancreas: A Focus on How Gut Microbiota Shapes Pancreatic Beta-Cells



Abstract
:1. Introduction
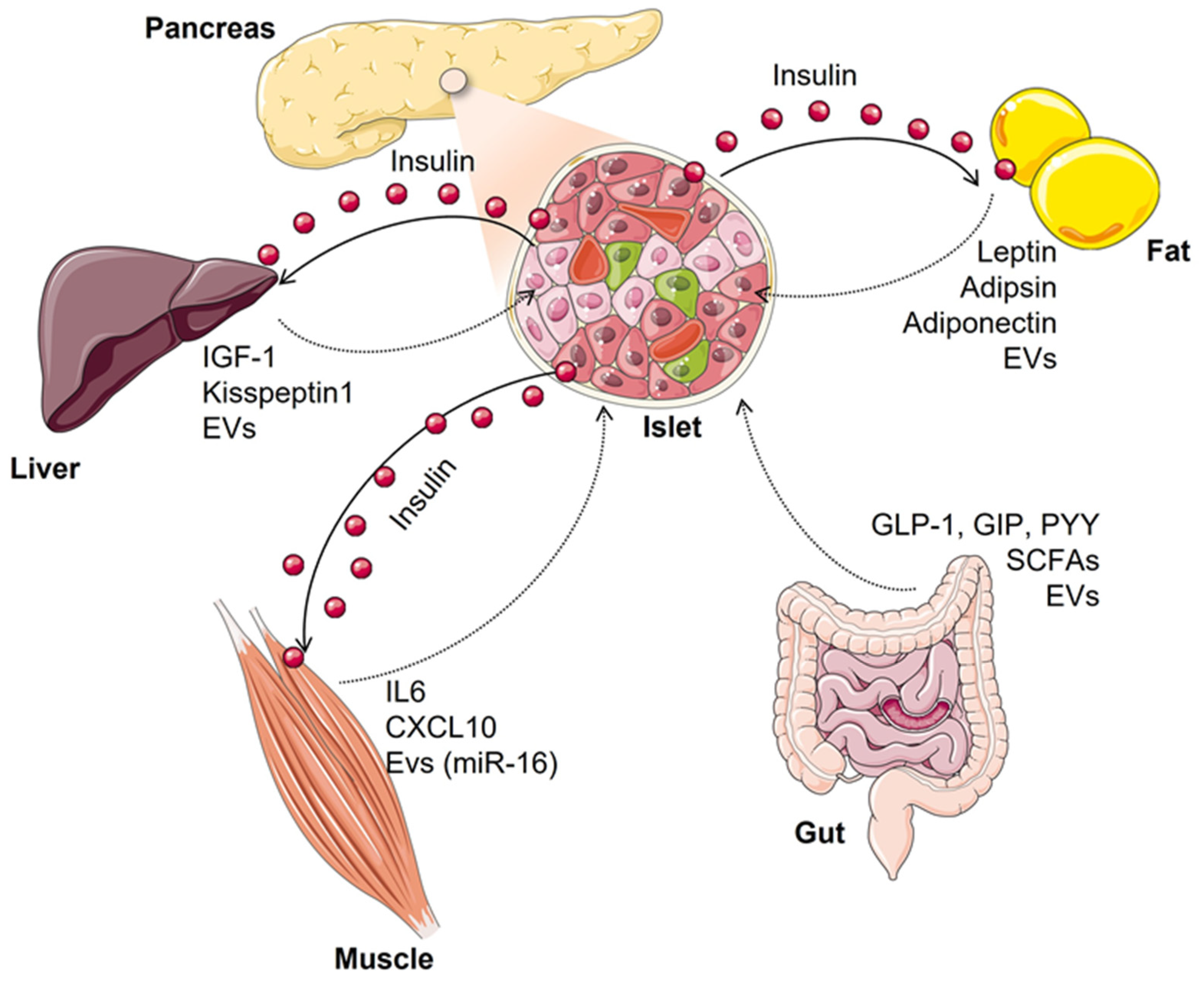
2. Inter-Organ Crosstalk Impacting Beta-Cell Function and Mass
2.1. Fat-Cell to Beta-Cell Communication
2.2. Hepatocyte to Beta-Cell Communication
2.3. Muscle-Cell to Beta-Cell Communication
2.4. Gut to Beta-Cell Communication
3. Diet Shapes Gut Microbiota
4. Gut Microbiota Shapes Pancreatic Beta-Cells
4.1. Modulation of Islet Responses by Short-Chain Fatty Acids (SCFAs)
4.2. Modulation of Islet Responses by Extracellular Vesicles (EVs)
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- IDF 10th Edition Diabetes Atlas. Available online: https://diabetesatlas.org/atlas/tenth-edition/ (accessed on 10 November 2021).
- Saisho, Y.; Butler, A.E.; Manesso, E.; Elashoff, D.; Rizza, R.A.; Butler, P.C. Beta-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Butler, A.E. Alterations in Beta Cell Identity in Type 1 and Type 2 Diabetes. Curr. Diab. Rep. 2019, 19, 83. [Google Scholar] [CrossRef] [Green Version]
- Cohrs, C.M.; Panzer, J.K.; Drotar, D.M.; Enos, S.J.; Kipke, N.; Chen, C.; Bozsak, R.; Schöniger, E.; Ehehalt, F.; Distler, M.; et al. Dysfunction of Persisting beta Cells Is a Key Feature of Early Type 2 Diabetes Pathogenesis. Cell Rep. 2020, 31, 107469. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of beta-cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontés, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.B.; Poliakov, A.; Hardy, R.W.; Clements, R.; Liu, C.; Liu, Y.; Wang, J.; Xiang, X.; Zhang, S.; Zhuang, X.; et al. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin reistance. Diabetes 2009, 58, 2498–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Sundaram, K.; Mu, J.; Dryden, G.W.; Sriwastva, M.K.; Lei, C.; Zhang, L.; Qiu, X.; Xu, F.; Yan, J.; et al. High-fat diet-induced upregulation of exosomal phosphatidylcholine contributes to insulin resistance. Nat. Commun. 2021, 12, 213. [Google Scholar] [CrossRef]
- Gesmundo, I.; Pardini, B.; Gargantini, E.; Gamba, G.; Birolo, G.; Fanciulli, A.; Banfi, D.; Congiusta, N.; Favaro, E.; Deregibus, M.C.; et al. Adipocyte-derived extracellular vesicles regulate survival and function of pancreatic β cells. JCI Insight 2021, 6, e141962. [Google Scholar] [CrossRef]
- Díez-Sainz, E.; Milagro, F.I.; Riezu-Boj, J.I.; Lorente-Cebrián, S. Effects of gut microbio-ta-derived extracellular vesicles on obesity and diabetes and their potential modula-tion through diet. J. Physiol. Biochem. 2021, 2, 1–15. [Google Scholar]
- Kobayashi, Y.; Eguchi, A.; Tempaku, M.; Honda, T.; Togashi, K.; Iwasa, M.; Hasegawa, H.; Takei, Y.; Sumida, Y.; Taguchi, O. Circulating extracellular vesicles are associated with lipid and insulin metabolism. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E574–E582. [Google Scholar] [CrossRef] [PubMed]
- Chidester, S.; Livinski, A.A.; Fish, A.F.; Joseph, P.V. The Role of Extracellular Vesicles in β-Cell Function and Viability: A Scoping Review. Front Endocrinol. 2020, 11, 375. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E. The many secret lives of adipocytes: Implications for diabetes. Diabetologia 2019, 62, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [Green Version]
- Kieffer, T.J.; Heller, R.S.; Habener, J.F. Leptin receptors expressed on pancreatic beta-cells. Biochem. Biophys. Res. Commun. 1996, 224, 522–527. [Google Scholar] [CrossRef]
- Wu, Y.; Fortin, D.A.; Cochrane, V.A.; Chen, P.C.; Shyng, S.L. NMDA receptors mediate leptin signaling and regulate potassium channel trafficking in pancreatic β-cells. J. Biol. Chem. 2017, 292, 15512–15524. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-C.; Kryukova, Y.N.; Shyng, S.-L. Leptin regulates KATP channel trafficking in pancreatic β-cells by a signaling mechanism involving AMP-activated protein kinase (AMPK) and cAMP-dependent protein kinase (PKA). J. Biol. Chem. 2013, 288, 34098–34109. [Google Scholar] [CrossRef] [Green Version]
- Laubner, K.; Kieffer, T.J.; Lam, N.T.; Niu, X.; Jakob, F.; Seufert, J. Inhibition of preproinsulin gene expression by leptin induction of suppressor of cytokine signaling 3 in pancreatic beta-cells. Diabetes 2005, 54, 3410–3417. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Banoy, N.; Guseh, J.S.; Li, G.; Rubio-Navarro, A.; Chen, T.; Poirier, B.; Putzel, G.; Rosselot, C.; Pabón, M.A.; Camporez, J.P.; et al. Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat. Med. 2019, 25, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Holland, W.L.; Gordillo, R.; Wang, M.; Wang, Q.A.; Shao, M.; Morley, T.S.; Gupta, R.K.; Stahl, A.; Scherer, P.E. Adiponectin is essential for lipid homeostasis and survival under insulin deficiency and promotes β-cell regeneration. ELife 2014, 3, e03851. [Google Scholar] [CrossRef] [PubMed]
- El Ouaamari, A.; Kawamori, D.; Dirice, E.; Liew, C.W.; Shadrach, J.L.; Hu, J.; Katsuta, H.; Hollister-Lock, J.; Qian, W.J.; Wagers, A.J.; et al. Liver-derived systemic factors drive β cell hyperplasia in insulin-resistant states. Cell Rep. 2013, 3, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Escribano, O.; Guillén, C.; Nevado, C.; Gómez-Hernández, A.; Kahn, C.R.; Benito, M. Beta-Cell hyperplasia induced by hepatic insulin resistance: Role of a liver-pancreas endocrine axis through insulin receptor A isoform. Diabetes 2009, 8, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Mondal, P.; Wolfe, A.; Alonso, L.C.; Stamateris, R.; Ong, B.W.; Lim, O.C.; Yang, K.S.; Radovick, S.; Novaira, H.J.; et al. Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab. 2014, 19, 667–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowe, J.E.; Hill, T.G.; Hunt, K.F.; Smith, L.I.; Simpson, S.J.; Amiel, S.A.; Jones, P.M. A role for placental kisspeptin in β cell adaptation to pregnancy. JCI Insight 2019, 4, e124540. [Google Scholar] [CrossRef]
- Fu, F.; Li, Y.; Jiang, H.; Shen, Z.; Gao, R.; He, Y.; Liu, Y.; Xu, K.; Yang, T. Hepatocytes derived extracellular vesicles from high-fat diet induced obese mice modulate genes expression and proliferation of islet β cells. Biochem. Biophys. Res. Commun. 2019, 516, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Hjorth, M.F.; Astrup, A. Diet and exercise in the prevention and treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Ge, L.; Xun, Y.-Q.; Chen, Y.-J.; Gao, C.-Y.; Han, X.; Zuo, L.-Q.; Shan, H.-Q.; Yang, K.-H.; Ding, G.-W.; et al. Exercise training modalities in patients with type 2 diabetes mellitus: A systematic review and network meta-analysis. Int. J. Behav. Nutr. Phys. Act. 2018, 15, 72. [Google Scholar] [CrossRef]
- Ellingsgaard, H.; Hauselmann, I.; Schuler, B.; Habib, A.M.; Baggio, L.L.; Meier, D.T.; Eppler, E.; Bouzakri, K.; Wueest, S.; Muller, Y.D.; et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Roytblat, L.; Rachinsky, M.; Fisher, A.; Greemberg, L.; Shapira, Y.; Douvdevani, A.; Gelman, S. Raised interleukin-6 levels in obese patients. Obes. Res. 2000, 8, 673–675. [Google Scholar] [CrossRef]
- Klover, P.J.; Zimmers, T.A.; Koniaris, L.G.; Mooney, R.A. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 2003, 52, 2784–2789. [Google Scholar] [CrossRef] [Green Version]
- Bouzakri, K.; Plomgaard, P.; Berney, T.; Donath, M.Y.; Pedersen, B.K.; Halban, P.A. Bimodal effect on pancreatic beta-cells of secretory products from normal or insulin-resistant human skeletal muscle. Diabetes 2011, 60, 1111–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, J.P.; Solomon, T.P. Do skeletal muscle-secreted factors influence the function of pancreatic β-cells? Am. J. Physiol. Endocrinol. Metab. 2018, 314, E297–E307. [Google Scholar] [CrossRef]
- Russell, A.P.; Lamon, S.; Boon, H.; Wada, S.; Güller, I.; Brown, E.L.; Chibalin, A.V.; Zierath, J.R.; Snow, R.J.; Stepto, N.I.; et al. Regulation of miRNAs in human skeletal muscle following acute endurance exercise and short-term endurance training. J. Physiol. 2013, 591, 4637–4653. [Google Scholar] [CrossRef] [PubMed]
- Karolina, D.S.; Armugam, A.; Tavintharan, S.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. MicroRNA 144 Impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE 2011, 6, e22839. [Google Scholar] [CrossRef]
- Jalabert, A.; Vial, G.; Guay, C.; Wiklander, O.P.; Nordin, J.Z.; Aswad, H.; Forterre, A.; Meugnier, E.; Pesenti, S.; Regazzi, R.; et al. Exosome-like vesicles released from lipid-induced insulin-resistant muscles modulate gene expression and proliferation of beta recipient cells in mice. Diabetologia 2016, 59, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Bayliss, W.M.; Starling, E.H. On the causation of the so-called ‘peripheral reflex secretion’ of the pancreas. Proc. R. Soc. Lond. Biol. 1902, 69, 352–353. [Google Scholar]
- Moore, B.; Edie, E.S.; Abram, J.H. On the treatment of diabetes mellitus by acid extract of duodenal mucous membrane. Biochem. J. 1906, 1, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Elrick, H.; Stimmler, L.; Hlad, C.J., Jr.; Arai, Y. Plasma insulin response to oral and intravenous glucose administration. J. Clin. Investig. 1964, 24, 1076–1082. [Google Scholar]
- Kreymann, B.; Williams, G.; Ghatei, M.A.; Bloom, S.R. Glucagon-like peptide-1 7-36: A physiological incretin in man. Lancet 1987, 2, 1300–1304. [Google Scholar] [CrossRef]
- Li, Y.; Cao, X.; Li, L.X.; Brubaker, P.L.; Edlund, H.; Drucker, D.J. Beta-cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes 2005, 54, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Hansotia, T.; Yusta, B.; Ris, F.; Halban, P.A.; Drucker, D.J. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J. Biol. Chem. 2003, 278, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved incretin activity of glucagonlike peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J. Mechanisms of Action and Therapeutic. Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, N.B.; Jacobsen, S.H.; Dirksen, C.; Bojsen-Møller, K.N.; Naver, L.P.S.; Hvolris, L.; Clausen, T.R.; Wulff, B.S.; Worm, D.; Hansen, D.L.; et al. Acute and long-term effects of Roux-en-Y gastric bypass on glucose metabolism in subjects with Type 2 diabetes and normal glucose tolerance. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E122–E131. [Google Scholar] [CrossRef] [Green Version]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Indias, I.; Cardona, F.; Tinahones, F.J.; Queipo-Ortuño, M.I. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol. 2014, 5, 190. [Google Scholar] [CrossRef] [Green Version]
- Salim, S.Y.; Söderholm, J.D. Importance of Disrupted Intestinal Barrier in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2011, 13, 62–381. [Google Scholar] [CrossRef]
- Al-Lahham, S.H.; Peppelenbosch, M.P.; Roelofsen, H.; Vonk, R.J.; Venema, K. Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochem. Biophys. Acta 2010, 1801, 1175–1183. [Google Scholar] [CrossRef]
- Delzenne, N.M.; Williams, C.M. Prebiotics and lipid metabolism. Curr. Opin. Lipidol. 2002, 13, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Freeland, K.R.; Wolever, T.M. Acute effects of intravenous and rectal acetate on glucagon-like peptide-1, peptide YY, ghrelin, adiponectin and tumour necrosis factor-alpha. Br. J. Nutr. 2010, 103, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Davis, B.; Zhu, W.; Zheng, N.; Meng, D.; Walker, W.A. Short-chain fatty acid butyrate, a breast milk metabolite, enhances immature intestinal barrier function genes in response to inflammation in vitro and in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G521–G530. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMPactivated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Martínez-Oca, P.; Robles-Vera, I.; Sánchez-Roncero, A.; Escrivá, F.; Pérez-Vizcaíno, F.; Duarte, J.; Álvarez, C.; Fernández-Millán, E. Gut dysbiosis and altered barrier function precedes the appearance of metabolic syndrome in a rat model of nutrient-induced catch-up growth. J. Nutr. Biochem. 2020, 81, 108383. [Google Scholar] [CrossRef]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Bäckhed, F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshini, M.; Wicksteed, B.; Schiltz, G.E.; Gilchrist, A.; Layden, B.T. SCFA Receptors in Pancreatic Beta Cells: Novel Diabetes Targets? Trends Endocrinol. Metab. 2016, 27, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Ahmed, K.; Gille, A.; Lu, S.; Gröne, H.J.; Tunaru, S.; Offermanns, S. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 2015, 21, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, A.; Chambers, E.S.; Hill, T.; Maldonado, I.R.; Liu, B.; Bewick, G.; Morrison, D.J.; Preston, T.; Wallis, G.A.; Tedford, C.; et al. The diet-derived short chain fatty acid propionate improves beta-cell function in humans and stimulates insulin secretion from human islets in vitro. Diabetes Obes. Metab. 2017, 19, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Pingitore, A.; Gonzalez-Abuin, N.; Ruz-Maldonado, I.; Huang, G.C.; Frost, G.; Persaud, S.J. Short chain fatty acids stimulate insulin secretion and reduce apoptosis in mouse and human islets in vitro: Role of free fatty acid receptor 2. Diabetes Obes. Metab. 2019, 21, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNelis, J.C.; Lee, Y.S.; Mayoral, R.; van der Kant, R.; Johnson, A.M.F.; Wollam, J.; Olefsky, J.M. GPR43 Potentiates β-Cell Function in Obesity. Diabetes 2015, 64, 3203–3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, Z.; Xiong, D.; Wu, M.; Ding, J.L. FFAR2-FFAR3 receptor heteromerization modulates short-chain fatty acid sensing. FASEB J. 2018, 32, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, S.; Nirmalkar, K.; Hoyo-Vadillo, C.; García-Espitia, M.; Ramírez-Sánchez, D.; García-Mena, J. Gut microbiome production of short-chain fatty acids and obesity in children. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 621–625. [Google Scholar] [CrossRef]
- González-Hernández, M.A.; Canfora, E.E.; Jocken, J.W.E.; Blaak, E.E. The Short-Chain Fatty Acid Acetate in Body Weight Control and Insulin Sensitivity. Nutrients 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, J.; Rani, K.; Datt, C. Molecular link between dietary fibre, gut microbiota and health. Mol. Biol. Rep. 2020, 47, 6229–6237. [Google Scholar] [CrossRef]
- Villa, S.R.; Priyadarshini, M.; Fuller, M.H.; Bhardwaj, T.; Brodsky, M.R.; Angueira, A.R.; Mosser, R.E.; Carboneau, B.A.; Tersey, S.A.; Mancebo, H.; et al. Loss of Free Fatty Acid Receptor 2 leads to impaired islet mass and beta cell survival. Sci. Rep. 2016, 6, 28159. [Google Scholar] [CrossRef] [Green Version]
- Figlia, G.; Willnow, P.; Teleman, A.A. Metabolites Regulate Cell Signaling and Growth via Covalent Modification of Proteins. Dev. Cell 2020, 54, 156–170. [Google Scholar] [CrossRef]
- Zhang, J.J.; Fan, T.T.; Mao, Y.Z.; Hou, J.L.; Wang, M.; Zhang, M.; Lin, Y.; Zhang, L.; Yan, G.Q.; An, Y.P.; et al. Nuclear dihydroxyacetone phosphate signals nutrient sufficiency and cell cycle phase to global histone acetylation. Nat. Metab. 2021, 3, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Park, S.J.; Lee, H.; Siddiqi, F.; Lee, J.E.; Menzies, F.M.; Rubinsztein, D.C. Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab. 2019, 29, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Aguilar, A.; Guillén, C.; Nellist, M.; Bartolomé, A.; Benito, M. TSC2 N-terminal lysine acetylation status affects to its stability modulating mTORC1 signaling and autophagy. BBA Mol. Cell Res. 2016, 1863, 2658–2667. [Google Scholar] [CrossRef]
- Hu, S.; Kuwabara, R.; de Haan, B.J.; Smink, A.M.; de Vos, P. Acetate and Butyrate Improve β-cell Metabolism and Mitochondrial Respiration under Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 1542. [Google Scholar] [CrossRef] [Green Version]
- Fellows, R.; Varga-Weisz, P. Chromatin dynamics and histone modifications in intestinal microbiota-host crosstalk. Mol. Metab. 2020, 38, 100925. [Google Scholar] [CrossRef]
- Krautkramer, K.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-L.; Segovia, I.; Yuan, X.-L.; Gao, Z.-H. Controversial Roles of Gut Microbiota-Derived Short-Chain Fatty Acids (SCFAs) on Pancreatic β-Cell Growth and Insulin Secretion. Int. J. Mol. Sci. 2020, 21, 910. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Watanabe, T.; Kamata, K.; Hara, A.; Minaga, K.; Kudo, M. Intestinal Dysbiosis and Autoimmune Pancreatitis. Front Immunol. 2021, 12, 621532. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Jobin, C. Microbiota in pancreatic health and disease: The next frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Li, L. Modulation of Short-Chain Fatty Acids as Potential Therapy Method for Type 2 Diabetes Mellitus. Can. J. Infect. Dis. Med. Microbiol. 2021, 2021, 6632266. [Google Scholar]
- Van de Wouw, M.; Boehme, M.; Lyte, J.M.; Wiley, N.; Strain, C.; O’Sullivan, O.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 2018, 596, 4923–4944. [Google Scholar] [CrossRef]
- Zhao, L.; Ye, Y.; Gu, L.; Jian, Z.; Stary, C.M.; Xiong, X. Extracellular vesicle-derived miRNA as a novel regulatory system for bi-directional communication in gut-brain-microbiota axis. J. Transl. Med. 2021, 19, 202. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. Synergistic Effects of Milk-Derived Exosomes and Galactose on α-Synuclein Pathology in Parkinson’s Disease and Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 1059. [Google Scholar] [CrossRef]
- Burillo, J.; Fernández-Rhodes, M.; Piquero, M.; López-Alvarado, P.; Menéndez, J.C.; Jiménez, B.; González-Blanco, C.; Marqués, P.; Guillén, C.; Benito, M. Human amylin aggregates release within exosomes as a protective mechanism in pancreatic β cells: Pancreatic β-hippocampal cell communication. BBA Mol. Cell Res. 2021, 1868, 118971. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garrido, N.; Cordero, C.; Olivo-Martinez, Y.; Badia, J.; Baldomà, L. Cell-to-Cell Communication by Host-Released Extracellular Vesicles in the Gut: Implications in Health and Disease. Int. J. Mol. Sci. 2021, 22, 2213. [Google Scholar] [CrossRef]
- Melnik, B.C.; Stremmel, W.; Weiskirchen, R.; John, S.M.; Schmitz, G. Exosome-Derived MicroRNAs of Human Milk and Their Effects on Infant Health and Development. Biomolecules 2021, 11, 851. [Google Scholar] [CrossRef]
- Dahiya, D.K.; Puniya, M.; Shandilya, U.K.; Dhewa, T.; Kumar, N.; Kumar, S.; Puniya, A.K.; Shukla, P. Gut Microbiota Modulation and Its Relationship with Obesity Using Prebiotic Fibers and Probiotics: A Review. Front Microbiol. 2017, 8, 563. [Google Scholar] [CrossRef]
- Ali, A.; Tan, H.; Kaiko, G.E. Role of the Intestinal Epithelium and Its Interaction with the Microbiota in Food Allergy. Front Immunol. 2020, 11, 604054. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Raposo, G.; Candalh, C.; Boussac, M.; Hershberg, R.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 2001, 121, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Sultan, S.; Mottawea, W.; Yeo, J.D.; Hammami, R. Gut Microbiota Extracellular Vesicles as Signaling Molecules Mediating Host-Microbiota Communications. Int. J. Mol. Sci. 2021, 22, 13166. [Google Scholar] [CrossRef]
- Díaz-Garrido, N.; Badia, J.; Baldomà, L. Microbiota-derived extracellular vesicles in interkingdom communication in the gut. J. Extracell. Vesicles 2021, 10, e12161. [Google Scholar] [CrossRef] [PubMed]
- Stentz, R.; Carvalho, A.L.; Jones, E.J.; Carding, S.R. Fantastic Voyage: The Journey of Intestinal Microbiota-Derived Microvesicles through the Body. Biochem. Soc. Transl. 2018, 46, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Luo, Z.; Jin, Z.; Ji, Y.; Ying, W. Adipose Tissue Macrophages Modulate Obesity-Associated β Cell Adaptations through Secreted miRNA-Containing Extracellular Vesicles. Cells 2021, 10, 2451. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Luo, Z.; Jin, Z.; Ji, Y.; Ying, W. Human Mesenchymal Stem Cell Derived Exosomes Alleviate Type 2 Diabetes Mellitus by Reversing Peripheral Insulin Resistance and Relieving β-Cell Destruction. ACS Nano 2018, 12, 7613–7628. [Google Scholar]
- Nojehdehi, S.; Soudi, S.; Hesampour, A.; Rasouli, S.; Soleimani, M.; Hashemi, S.M. Immunomodulatory effects of mesenchymal stem cell–derived exosomes on experimental type-1 autoimmune diabetes. J. Cell Biochem. 2018, 119, 9433–9443. [Google Scholar] [CrossRef]
- Nojehdehi, S.; Soudi, S.; Hesampour, A.; Rasouli, S.; Soleimani, M.; Hashemi, S.M. MicroRNAs 106b and 222 Improve Hyperglycemia in a Mouse Model of Insulin-Deficient Diabetes via Pancreatic β-Cell Proliferation. EBioMedicine 2017, 15, 163–172. [Google Scholar]
- Li, Y.; Deng, S.; Peng, J.; Wang, X.; Essandoh, K.; Mu, X.; Peng, T.; Meng, Z.-X.; Fan, G.-C. MicroRNA-223 is essential for maintaining functional β-cell mass during diabetes through inhibiting both FOXO1 and SOX6 pathways. J. Biol. Chem. 2019, 294, 10438–10448. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Kruit, J.K.; Rome, S.; Menoud, V.; Mulder, N.L.; Jurdzinski, A.; Mancarella, F.; Sebastiani, G.; Donda, A.; Gonzalez, B.J.; et al. Lymphocyte-Derived Exosomal MicroRNAs Promote Pancreatic β Cell Death and May Contribute to Type 1 Diabetes Development. Cell Metab. 2019, 29, 348–361. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Tissue Origin | Model | Cargo | Effect on Pancreatic β Cells | Reference |
|---|---|---|---|---|
| Adipose tissue macrophages | Obesity in vitro and in vivo | miR-155 | Increased in proliferation Decreased in insulin secretion | [95] |
| Adipocytes | Inflammation | Multiple miRNAs | Decreased cell survival Decreased insulin secretion | [13] |
| Mesenchymal stem cells (human umbilical cord) | T2DM | Not determined | Reversion of insulin resistance Decrease in pancreatic β cell death | [96] |
| Mesenchymal stem cells derived from adipose tissue | T2DM | Not determined | Increased β cell viability | [97] |
| Bone marrow | Hyperglycemia by STZ injection | miR106b-5p miR-222-3p | Promote pancreatic β cell proliferation | [98] |
| Skeletal muscle | T2DM | miR-16 | Increased pancreatic β cell proliferation Increased in insulin secretion | [40] |
| Hepatocytes | Obesity (T2DM) | miR-7218-5p | Increased in pancreatic β cell proliferation | [29] |
| Not determined | STZ-treated animals+/−HFD | miR-223 | Increased in pancreatic β cell proliferation Increased in insulin secretion Decreased β cell apoptosis | [99] |
| T-lymphocytes | T1DM | miR-142-3p miR-142-5p miR-155 | Stimulates pancreatic β cell apoptosis | [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Millán, E.; Guillén, C. Multi-Organ Crosstalk with Endocrine Pancreas: A Focus on How Gut Microbiota Shapes Pancreatic Beta-Cells. Biomolecules 2022, 12, 104. https://doi.org/10.3390/biom12010104
Fernández-Millán E, Guillén C. Multi-Organ Crosstalk with Endocrine Pancreas: A Focus on How Gut Microbiota Shapes Pancreatic Beta-Cells. Biomolecules. 2022; 12(1):104. https://doi.org/10.3390/biom12010104
Chicago/Turabian StyleFernández-Millán, Elisa, and Carlos Guillén. 2022. "Multi-Organ Crosstalk with Endocrine Pancreas: A Focus on How Gut Microbiota Shapes Pancreatic Beta-Cells" Biomolecules 12, no. 1: 104. https://doi.org/10.3390/biom12010104
APA StyleFernández-Millán, E., & Guillén, C. (2022). Multi-Organ Crosstalk with Endocrine Pancreas: A Focus on How Gut Microbiota Shapes Pancreatic Beta-Cells. Biomolecules, 12(1), 104. https://doi.org/10.3390/biom12010104