1. Introduction
Communities are significant components that are found in networks, such as social networks and biological networks. Its constituent elements are highly interconnected to perform the intended function. The structural, as well as functional, aspects of biological systems are well represented as biological networks comprising of different biomolecular elements. These networks can encode knowledge about local molecular interaction as well as some higher-level cellular communication. Studies show that changes to the network properties are very much linked to the phenotypes, such as tumors and mendelian disorders [
1]. Network data in the form of interactome, functional regulatory networks and gene co-expression networks, along with other public repositories, helped biologists to gain a deep understanding of variations in cellular processes. The healthy condition of human beings can be considered the result of the perfect functioning of biological networks. While investigating the mechanism of diseases, it has been found that diverse causes of complex diseases act together to dys-regulate the same components of the cellular system [
2].
Consequently, the network biology approach has emerged as an effective approach for understanding the underlying mechanism of complex diseases, including cancer [
3]. In various cancer types, the disease condition is reflected through the perturbed state in pathways or molecular subnetworks [
4,
5]. Subnetworks are a collection of inter-connected molecules that perform a particular function. Finding dys-regulated subnetworks will help extracting useful biological information. Furthermore, mapping molecular expression data with protein interaction networks is found to be an efficient approach for effectively elucidating patterns from the network [
6]. The integrative approach of combining gene expression data with other biomolecular networks was found to be efficient in extracting disease phenotypes [
7]. An individual-specific network was constructed using gene expression correlations and protein–protein interactions (PPI). Here, the interacting genes in the network were found to be associated with disease states. Also, this approach could find some proteins linked to diseases that act as potential therapeutic targets [
8]. In the target-centric method of drug discovery, a single target approach fails in complex disease scenario due to drug resistance and other facts [
9,
10].
Synergistic drug combination therapy has become a new trend, targeting pathways and modules consisting of multiple prominent targets. During the paradigm shift happening in drug discovery through systems-level target focusing, mining such pathway-based drug targets became challenging. This paper concentrates more on investigating network-oriented targets that could supplement the synergistic drugs in combating complex diseases. Thus, mining of dys-regulated subnetworks in multi-omics data has gained significance in drug design as well [
11].
During the past few decades, developing methods for extracting disease-related modules in molecular data was one of the major goals in computational biology. A variety of approaches have been applied to this computationally complex problem. The greedy approach, random walk, evolutionary approach and maximum clique identification are a few well-known methods, among others. Greedy methods such as Module Analysis via Topology of Interactions and Similarity Sets (MATISSE) and DIAMOnD employ seed molecule selection followed by expansion to derive disease modules [
12,
13]. Starting from the seed genes, neighbouring nodes are explored based on connectivity significance [
13]. Though the resulting disease modules are validated biologically, the greedy approaches fail to find optimum global networks. Another greedy method based on multivariate analysis used the scoring technique to derive a differentially expressed subnetwork [
14]. As these approaches are developed based only on exploitation to construct the path, a global optimum solution is not guaranteed.
GLADIATOR is an algorithm that made use of the evolutionary global search approach to derive disease modules. A simulated annealing algorithm is applied here to maximise the gold standard module similarity measure [
15]. Unlike other evolutionary algorithms, it uses a similarity index concerning known disease modules as an objective function. Moreover, it does not perform any statistical evaluation of the obtained results. However, the pure evolutionary algorithms require a precise objective function to measure actual perturbation in the module. HotNet2 is another algorithm developed for finding cancer subnetworks by mapping the connection strength to heat diffused over the network links [
16]. EnrichNet is a random walk approach associated with restart ability to identify known subnetworks that are strongly connected to input genes [
17].
Walktrap-GM is another algorithm that follows a random walk, exploiting the neighbours through the transition probability assessment on the weight value. A merge process of selected communities was also done to maximise the network modularity. Though this approach finds cancer-relevant modules, due to community-related computations, the complexity becomes O(n
3) for sparse data [
18]. A multi-objective approach is implemented, combining the properties, including module scores from gene expression, the pathway coverage score and connectivity measure [
19]. Although prior information regarding pathway enrichment is incorporated into the algorithm to extract active modules, the drug-related functionality analysis is not provided. Breast cancer modules were generated by IODNE by running a modified minimum spanning tree algorithm upon gene-protein data. This approach has extracted dys-regulated modules with the presence of a few drug targets. However, no statistical analysis has been conducted to validate the retrieved modules [
20].
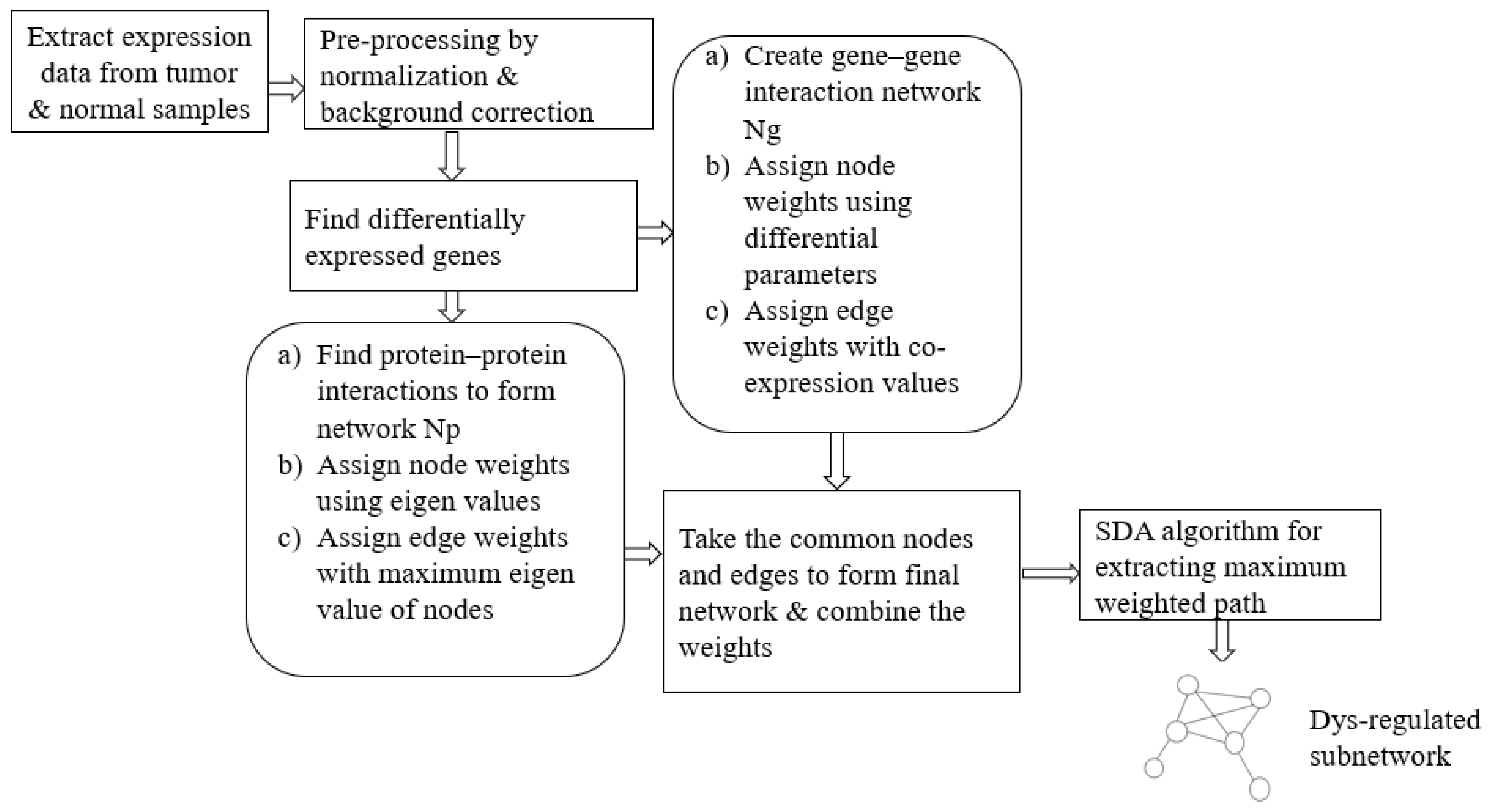
In our approach, we propose an ensemble of nature-inspired greedy approaches where the algorithm complexity is reduced. Most of the existing approaches initiate the search process from genes that are found to be relevant either topologically or biologically. Moreover, these methods suffer from extensive computations in the form of the repeated objective value calculation. In the proposed algorithm, the searching is performed by multiple agents starting from random nodes in the network and hence avoids the necessity for any prioritisation of start nodes. Additionally, the algorithm complexity has been reduced over existing greedy approaches.
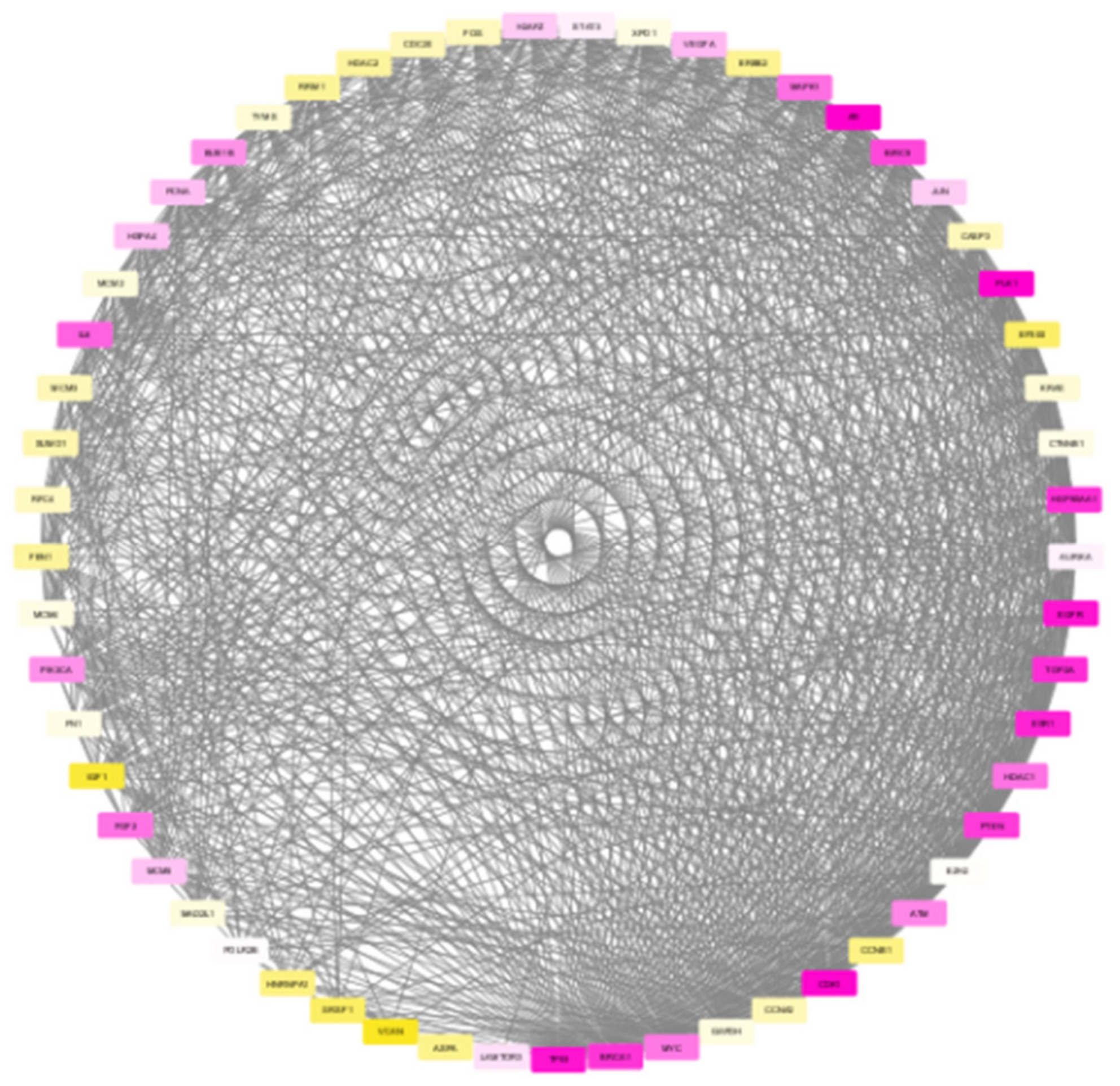
To test the proposed framework, gene expression data of the two most aggressive types of cancers, which affect the female and male category, were considered. Triple-negative breast cancer (TNBC) and colorectal cancer (CRC) samples were taken to generate the weighted network and for the subsequent subnetwork finding.
4. Limitations and Future Work
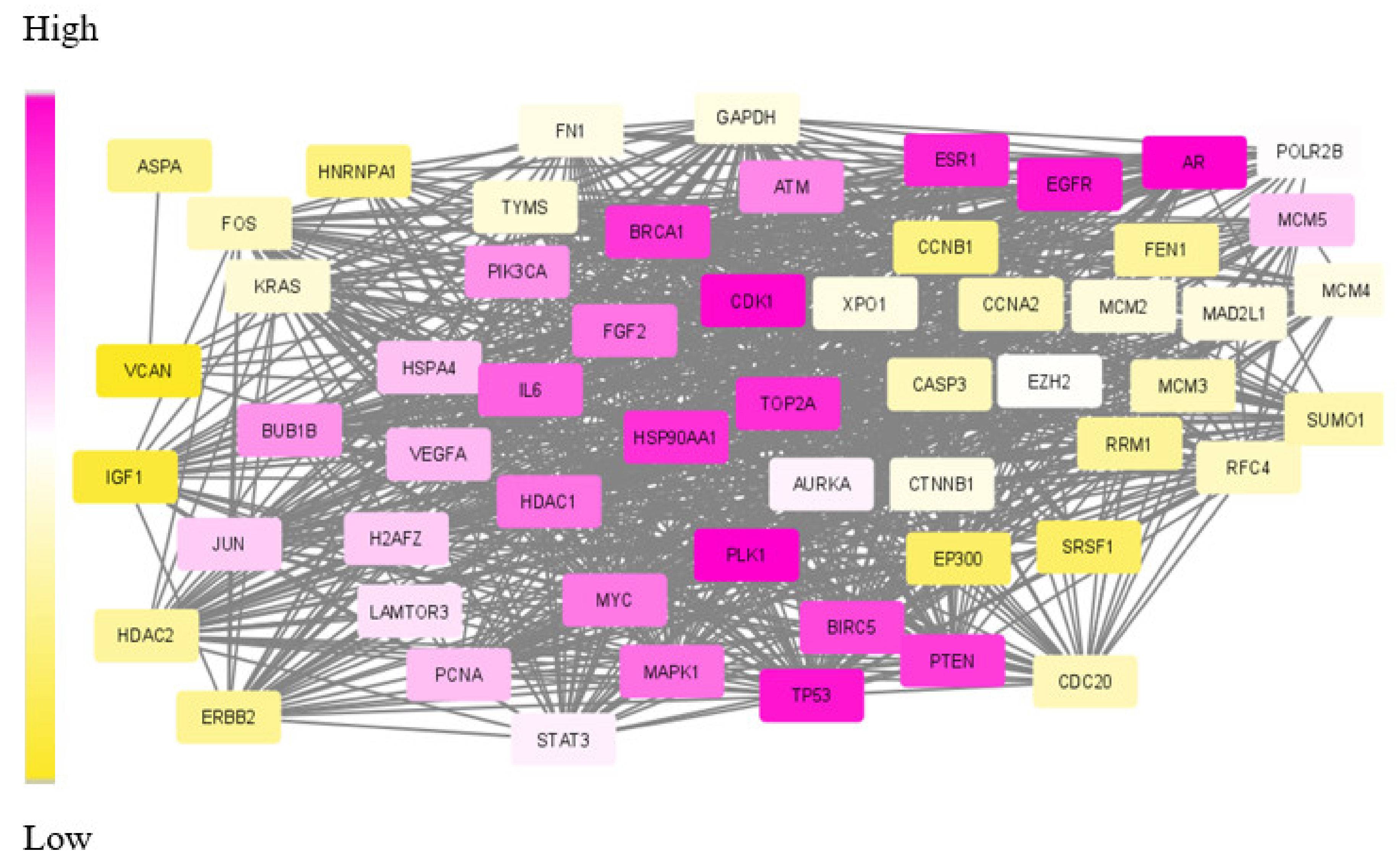
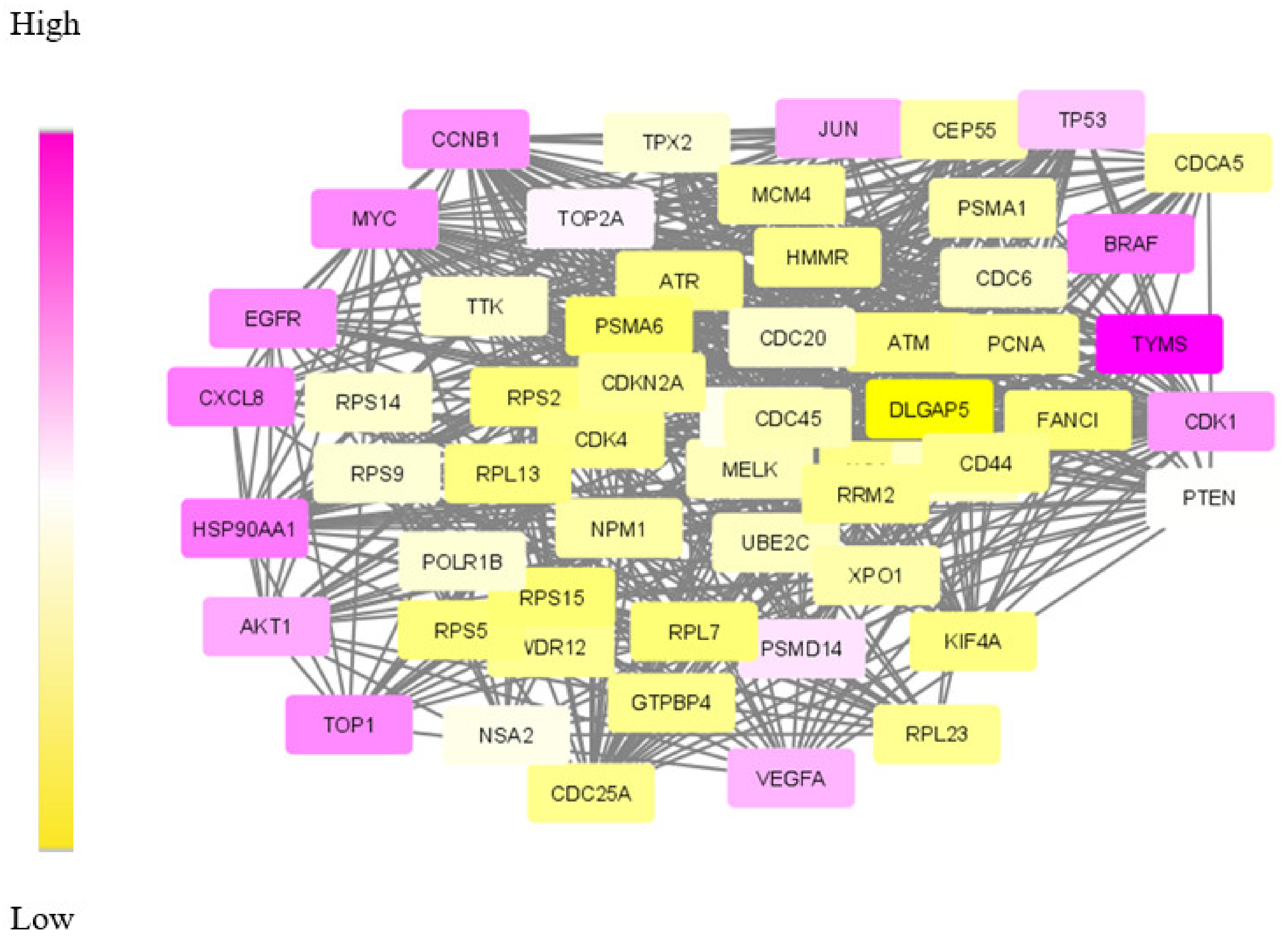
Our proposed approach has succeeded in extracting the de-regulated subnetwork in both TNBC and CRC data. In TNBC data, we could detect relevant target proteins, including proteins for synergistic drugs. While analysing the CRC module, we could find a couple of disease biomarkers and drug targets. However, in the data modules identified, our approach failed to identify some particular marker genes. KRAS and PAICS are two significant genes in CRC, but these were not included in the derived module. This may be due to the limited number of data samples taken for analysis.
Furthermore, we have considered microarray expression data for the analysis. With the advancements in sequencing technologies, RNA-Seq data sets are currently available for analysis. We could not use these transcriptome data for this study due to some technical constraints. However, our future work would concentrate on extracting RNA-seq data of cancer samples so as to derive more accurate results.
Similarly, the limitation with the smaller number of samples would be overcome by extracting more data samples. Additionally, the differential expression analysis would be performed by highly sophisticated methods. The final DE gene list would be prepared by taking the common genes obtained by each data sample. This is expected to improve the confidence of the initial seed genes for further analysis.
Another aspect of the de-regulated module that can be considered is the copy number variation count. Combining these three attributes would make the tool much more effective in module extraction.
5. Conclusions
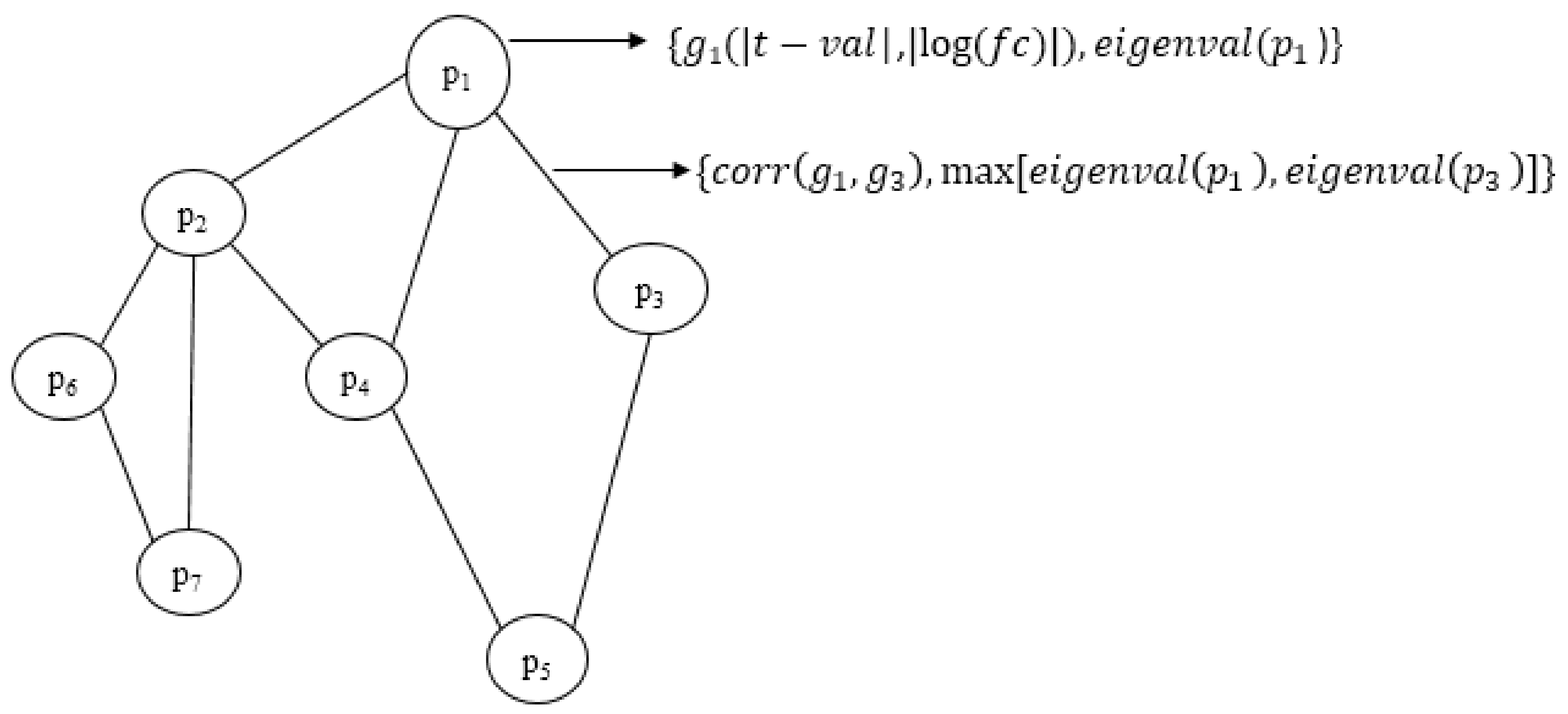
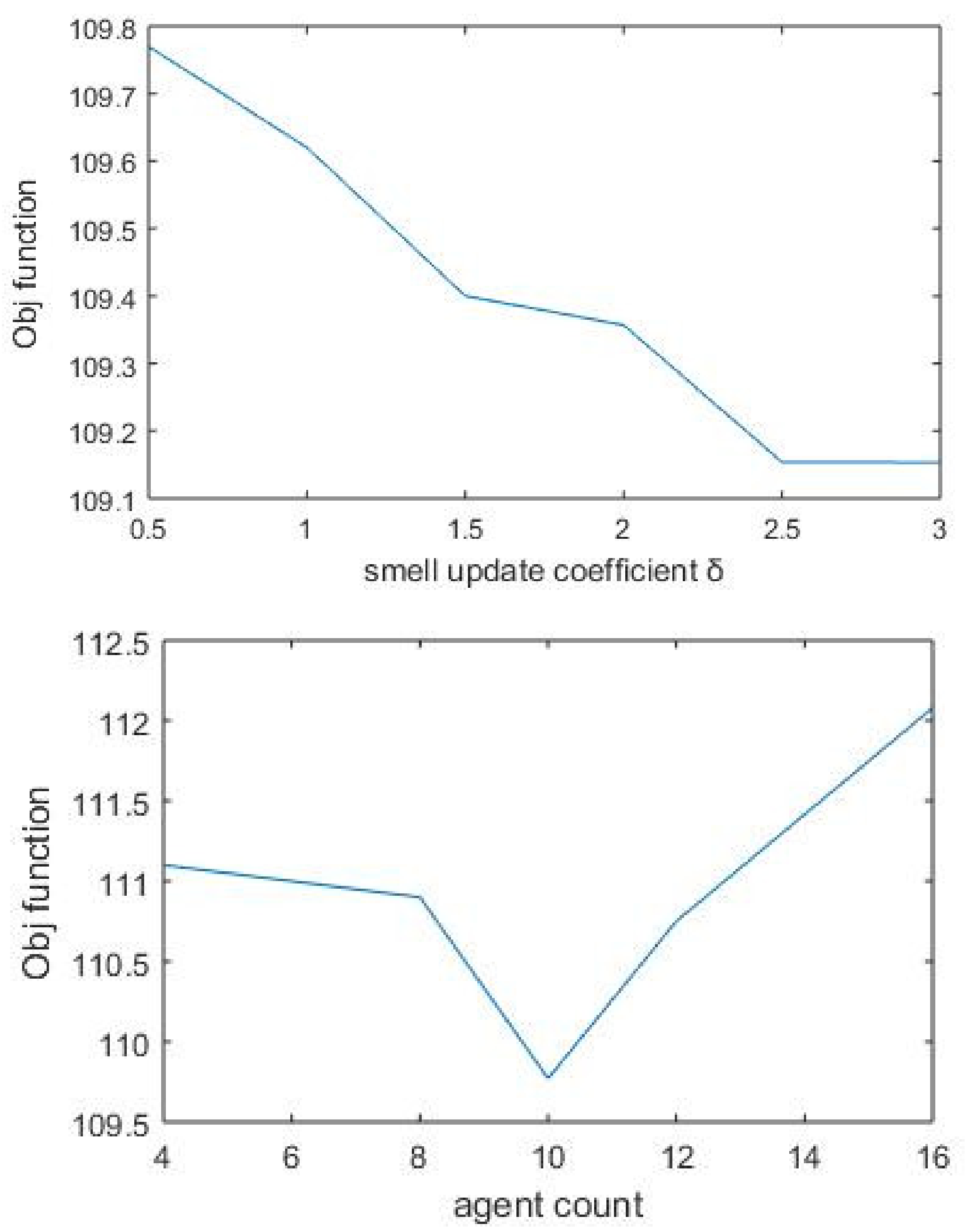
We have proposed an optimisation framework to elucidate the dys-regulated subnetwork from a weighted network curated out of differentially expressed genes and the corresponding proteins. An efficient nature-inspired SDA algorithm was designed for this path extraction. The most promising feature of this algorithm was the reduced time complexity of n (log n) for n number of nodes in the network. This algorithm has successfully derived the most optimum set of nodes and links based on the topological and differential expression scores. As we provided multiple agents, the algorithm has chosen the best path as the final result. These nodes were mapped to genes/proteins to form the molecular subnetwork to extract maximum biological information. Once we can extract such modules, we can process it further to mine useful information.
The biological evaluation of the obtained genes in the module has revealed the efficacy of our proposed approach. Due to the deadly nature and higher death rates, we have chosen TNBC and CRC data sets for analysis. Overall, in both these cancer types, 70% of the genes were biologically validated, including drug target prediction. In CRC, we proposed new drug targets considering the significance of the genes in the derived module.
Compared to the other approaches, the major advantage is that a single algorithm is sufficient to elucidate the module comprising of biomarkers, hub genes, drug targets, and other aspects. In most of the existing approaches, multiple tools and techniques are required to obtain all this information.
Above all, these modules’ future applications can be further analysed to access synergistic drug targets for the concerned disease. Through effective mechanisms, the synergistic targets which are likely to be bound by multiple drugs or small molecules can be recognised.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}